Molecular Regulation of the RhoGAP GRAF3 and Its Capacity to Limit Blood Pressure In Vivo


Abstract
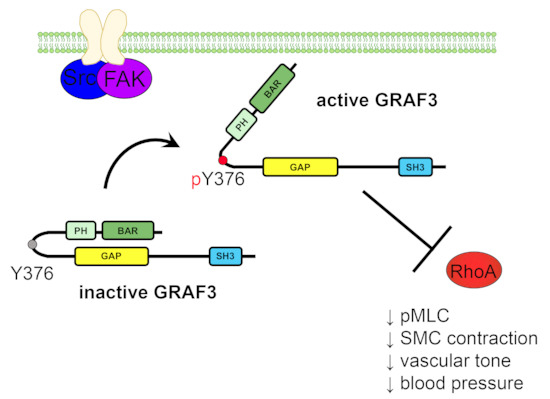
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation and Characterization of GRAF3RQ SMMHC-CreERT2 Mice
2.2. Blood Pressure Measurements
2.3. Cell Culture
2.4. Molecular Modeling
2.5. FRET Conformation Assay
2.6. Western Blotting
2.7. Immunoprecipitation
2.8. Protein Purification
2.9. Radioactive In Vitro Kinase assay
2.10. Time Resolved-Fluorescence Energy Transfer (TR-FRET) Assay
2.11. Bioluminescence Resonance Energy Transfer (BRET) Assay
2.12. Statistics
3. Results
3.1. Increased SMC GRAF3 Expression Reduced BP in Mice
3.2. GRAF3 Activity is Dynamically Regulated by Auto-Inhibitory Interactions
3.3. FAK and Src-Mediated Phosphorylation of GRAF3 at Y376 Promotes Allosteric Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nwankwo, T.; Yoon, S.S.; Burt, V.; Gu, Q. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011–2012. NCHS Data Brief. 2013, 133, 1–8. [Google Scholar]
- Achelrod, D.; Wenzel, U.; Frey, S. Systematic review and meta-analysis of the prevalence of resistant hypertension in treated hypertensive populations. Am. J. Hypertens. 2015, 28, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persell, S.D. Prevalence of resistant hypertension in the United States, 2003-2008. Hypertension 2011, 57, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farley, T.A.; Dalal, M.A.; Mostashari, F.; Frieden, T.R. Deaths Preventable in the U.S. by Improvements in Use of Clinical Preventive Services. Am. J. Prev. Med. 2010, 38, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W., Jr. The genetic dissection of essential hypertension. Nat. Rev. Genet. 2006, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Wu, X.; Nurkiewicz, T.R.; Kawasaki, J.; Davis, G.E.; Hill, M.A.; Meininger, G.A. Integrins and mechanotransduction of the vascular myogenic response. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1427–H1433. [Google Scholar] [CrossRef]
- Hall, J.E. The kidney, hypertension and obesity. Hypertension 2003, 41, 625–633. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Budzyn, K.; Marley, P.D.; Sobey, C.G. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharm. Sci. 2006, 27, 97–104. [Google Scholar] [CrossRef]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho- kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Loirand, G.; Pacaud, P. The role of Rho protein signaling in hypertension. Nat. Rev. Cardiol. 2010, 7, 637–647. [Google Scholar] [CrossRef]
- Mueller, B.K.; Mack, H.; Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, A. Rho kinase and hypertension. Biochim. Biophys. Acta. 2010, 1802, 1276–1284. [Google Scholar] [CrossRef]
- Masumoto, A.; Hirooka, Y.; Shimokawa, H.; Hironaga, K.; Setoguchi, S.; Takeshita, A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001, 38, 1307–1310. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Lenhart, K.C.; Bird, K.E.; Suen, A.A.; Rojas, M.; Kakoki, M.; Li, F.; Smithies, O.; Mack, C.P.; Taylor, J.M. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat. Commun. 2013, 4, 2910. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Mangum, K.D.; Dee, R.A.; Stouffer, G.A.; Lee, C.R.; Oni-Orisan, A.; Patterson, C.; Schisler, J.C.; Viera, A.J.; Taylor, J.M.; et al. Blood pressure–associated polymorphism controls ARHGAP42 expression via serum response factor DNA binding. J. Clin. Investig. 2017, 127, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; He, J.; Chen, J.; Zhao, J.; Gu, D.; Hixson, J.E.; Rao, D.C.; Jaquish, C.E.; Gu, C.C.; Chen, J.; et al. Genome-Wide Gene-Sodium Interaction Analyses on Blood Pressure: The Genetic Epidemiology Network of Salt-Sensitivity Study. Hypertension 2016, 68, 348–355. [Google Scholar] [CrossRef] [Green Version]
- The International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wain, L.V.; Verwoert, G.C.; O’Reilly, P.F.; Shi, G.; Johnson, T.; Johnson, A.D.; Bochud, M.; Rice, K.M.; Henneman, P.; Smith, A.V.; et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat. Genet. 2011, 43, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Fjorder, A.S.; Rasmussen, M.B.; Mehrjouy, M.B.; Nazaryan-Petersen, L.; Hansen, C.; Bak, M.; Grarup, N.; Norremolle, A.; Larsen, L.A.; Vestergaard, H.; et al. Haploinsufficiency of Arhgap42 is associated with hypertension. Eur. J. Hum. Genet. 2019, 27, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- DiMichele, L.A.; Hakim, Z.S.; Sayers, R.L.; Rojas, M.; Schwartz, R.J.; Mack, C.P.; Taylor, J.M. Transient expression of FRNK reveals stage-specific requirement for focal adhesion kinase activity in cardiac growth. Circ. Res. 2009, 104, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.M.T.; Lenhart, K.C.; Cameron, M.V.; Mack, C.P.; Conlon, F.L.; Taylor, J.M. Skeletal muscle differentiation and fusion are regulated by the BAR-containing Rho-GTPase-activating protein (Rho-GAP), GRAF1. J. Biol Chem 2011, 286, 25903–25921. [Google Scholar] [CrossRef] [Green Version]
- Gamblin, S.J.; Smerdon, S.J. GTPase-activating proteins and their complexes. Curr. Opin. Struct. Biol. 1998, 8, 195–201. [Google Scholar] [CrossRef]
- Longenecker, K.L.; Zhang, B.; Derewenda, U.; Sheffield, P.J.; Dauter, Z.; Parsons, J.M.T.; Zheng, Y.; Derewenda, Z.S. Structure of the BH domain from graf and its implications for Rho GTPase recognition. J. Biol. Chem. 2000, 275, 38605–38610. [Google Scholar] [CrossRef] [Green Version]
- Staus, D.P.; Blaker, A.L.; Taylor, J.M.; Mack, C.P. Diaphanous 1 and 2 regulate smooth muscle cell differentiation by activating the myocardin-related transcription factors. Arter. Thromb. Vasc. Biol. 2007, 27, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinson, J.S.; Medlin, M.D.; Lockman, K.; Taylor, J.M.; Mack, C.P. Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1170–H1180. [Google Scholar] [CrossRef] [PubMed]
- Lockman, K.; Hinson, J.S.; Medlin, M.D.; Morris, D.; Taylor, J.M.; Mack, C.P. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J. Biol. Chem. 2004, 279, 42422–42430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dee, R.A.; Mangum, K.D.; Bai, X.; Mack, C.P.; Taylor, J.M. Druggable targets in the Rho pathway and their promise for therapeutic control of blood pressure. Pharm 2019, 193, 121–134. [Google Scholar] [CrossRef]
- Eberth, A.; Lundmark, R.; Gremer, L.; Dvorsky, R.; Koessmeier, K.T.; McMahon, H.T.; Ahmadian, M.R. A BAR domain-mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem. J. 2009, 417, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Mao, X.; Dong, L.Q.; Liu, F.; Tong, L. Crystal structures of the BAR-PH and PTB domains of human APPL1. Structure 2007, 15, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Chen, J.; Liu, J.; Brunzelle, J.S.; Huang, B.; Wakeham, N.; Terzyan, S.; Li, X.; Rao, Z.; Li, G.; et al. Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5. EMBO J. 2007, 26, 3484–3493. [Google Scholar] [CrossRef] [Green Version]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arter. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Janostiak, R.; Tolde, O.; Ryzhova, L.M.; Koudelkova, L.; Dibus, M.; Brabek, J.; Hanks, S.K.; Rosel, D. ARHGAP42 is activated by Src-mediated tyrosine phosphorylation to promote cell motility. J. Cell Sci. 2017, 130, 2382–2393. [Google Scholar] [CrossRef] [Green Version]
- Gabarra-Niecko, V.; Keely, P.J.; Schaller, M.D. Characterization of an activated mutant of focal adhesion kinase: ‘SuperFAK’. Biochem. J. 2002, 365, 591–603. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.D.; Kiosses, W.B.; Schwartz, M.A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999, 18, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar]
- Terrell, E.M.; Durrant, D.E.; Ritt, D.A.; Sealover, N.E.; Sheffels, E.; Spencer-Smith, R.; Esposito, D.; Zhou, Y.; Hancock, J.F.; Kortum, R.L.; et al. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 2019, 76, 872–884 e875. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Hill, M.A. Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 1999, 79, 387–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Mangum, K.; Kakoki, M.; Smithies, O.; Mack, C.P.; Taylor, J.M. GRAF3 serves as a blood volume-sensitive rheostat to control smooth muscle contractility and blood pressure. Small Gtpases 2017. [Google Scholar] [CrossRef]
- Mangum, K.D.; Freeman, E.J.; Magin, J.C.; Taylor, J.; Mack, C.P. Transcriptional and post-transcriptional regulation of the blood pressure-associated Rho-specific GAP, ARHGAP42. Am. J. Physio. Heart Circ. Physio. 1984, 38, 745–755. [Google Scholar] [CrossRef]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Harst, P.v.d.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef]
- El-Yazbi, A.F.; Abd-Elrahman, K.S. ROK and Arteriolar Myogenic Tone Generation: Molecular Evidence in Health and Disease. Front. Pharm. 2017, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.L.; Bregeon, J.; Devos, N.; Chadeuf, G.; Blanchard, A.; Azizi, M.; Pacaud, P.; Jeunemaitre, X.; Loirand, G. Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertension 2015, 65, 1273–1278. [Google Scholar] [CrossRef] [Green Version]
- Guilluy, C.; Bregeon, J.; Toumaniantz, G.; Rolli-Derkinderen, M.; Retailleau, K.; Loufrani, L.; Henrion, D.; Scalbert, E.; Bril, A.; Torres, R.M.; et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 2010, 16, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P. Statin-mediated inhibition of Rho: Only to get more NO? Circ. Res. 2005, 96, 927–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaki, A.I.; Sarafidis, P.A.; Georgianos, P.I.; Kanavos, K.; Tziolas, I.M.; Zebekakis, P.E.; Lasaridis, A.N. Effects of low-dose atorvastatin on arterial stiffness and central aortic pressure augmentation in patients with hypertension and hypercholesterolemia. Am. J. Hypertens. 2013, 26, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumiya, S.; Thumkeo, D. Rho signaling research: History, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surma, M.; Wei, L.; Shi, J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011, 7, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef]
- Loirand, G.; Pacaud, P. Involvement of Rho GTPases and their regulators in the pathogenesis of hypertension. Small Gtpases 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Nishiki, K.; Tsuruoka, S.; Kawaguchi, A.; Sugimoto, K.; Schwartz, G.J.; Suzuki, M.; Imai, M.; Fujimura, A. Inhibition of Rho-kinase reduces renal Na-H exchanger activity and causes natriuresis in rat. J. Pharm. Exp. 2003, 304, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Holderfield, M. Efforts to Develop KRAS Inhibitors. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Canagarajah, B.; Leskow, F.C.; Ho, J.Y.; Mischak, H.; Saidi, L.F.; Kazanietz, M.G.; Hurley, J.H. Structural mechanism for lipid activation of the Rac-specific GAP, beta2-chimaerin. Cell 2004, 119, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskwa, P.; Paclet, M.H.; Dagher, M.C.; Ligeti, E. Autoinhibition of p50 Rho GTPase-activating protein (GAP) is released by prenylated small GTPases. J. Biol. Chem. 2005, 280, 6716–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, B.K.; Qian, X.; Mertins, P.; Wang, D.; Papageorge, A.G.; Carr, S.A.; Lowy, D.R. CDK5 is a major regulator of the tumor suppressor DLC1. J. Cell Biol. 2014, 207, 627–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, X.; Brown, P.; Schuck, P.; Gruschus, J.M.; Balbo, A.; Hinshaw, J.E.; Randazzo, P.A. Autoinhibition of Arf GTPase-activating protein activity by the BAR domain in ASAP1. J. Biol. Chem. 2009, 284, 1652–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, Y.; Ma, Q.; Vahedi-Faridi, A.; Sundborger, A.; Pechstein, A.; Puchkov, D.; Luo, L.; Shupliakov, O.; Saenger, W.; Haucke, V. Molecular basis for SH3 domain regulation of F-BAR–mediated membrane deformation. Proc. Natl. Acad. Sci. 2010, 107, 8213–8218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanishneva-Konovalova, T.B.; Derkacheva, N.I.; Polevova, S.V.; Sokolova, O.S. The Role of BAR Domain Proteins in the Regulation of Membrane Dynamics. Acta Nat. 2016, 8, 60–69. [Google Scholar] [CrossRef]
- Vázquez, F.X.; Unger, V.M.; Voth, G.A. Autoinhibition of endophilin in solution via interdomain interactions. Biophys. J. 2013, 104, 396–403. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dee, R.A.; Bai, X.; Mack, C.P.; Taylor, J.M. Molecular Regulation of the RhoGAP GRAF3 and Its Capacity to Limit Blood Pressure In Vivo. Cells 2020, 9, 1042. https://doi.org/10.3390/cells9041042
Dee RA, Bai X, Mack CP, Taylor JM. Molecular Regulation of the RhoGAP GRAF3 and Its Capacity to Limit Blood Pressure In Vivo. Cells. 2020; 9(4):1042. https://doi.org/10.3390/cells9041042
Chicago/Turabian StyleDee, Rachel A., Xue Bai, Christopher P. Mack, and Joan M. Taylor. 2020. "Molecular Regulation of the RhoGAP GRAF3 and Its Capacity to Limit Blood Pressure In Vivo" Cells 9, no. 4: 1042. https://doi.org/10.3390/cells9041042
APA StyleDee, R. A., Bai, X., Mack, C. P., & Taylor, J. M. (2020). Molecular Regulation of the RhoGAP GRAF3 and Its Capacity to Limit Blood Pressure In Vivo. Cells, 9(4), 1042. https://doi.org/10.3390/cells9041042