Breakthrough Technologies Reshape the Ewing Sarcoma Molecular Landscape

 and
and 
Abstract
: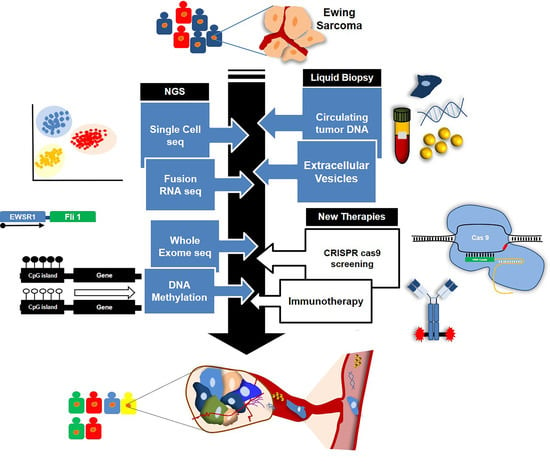
{kind=link}
{kind=link}
{kind=link}
1. Introduction
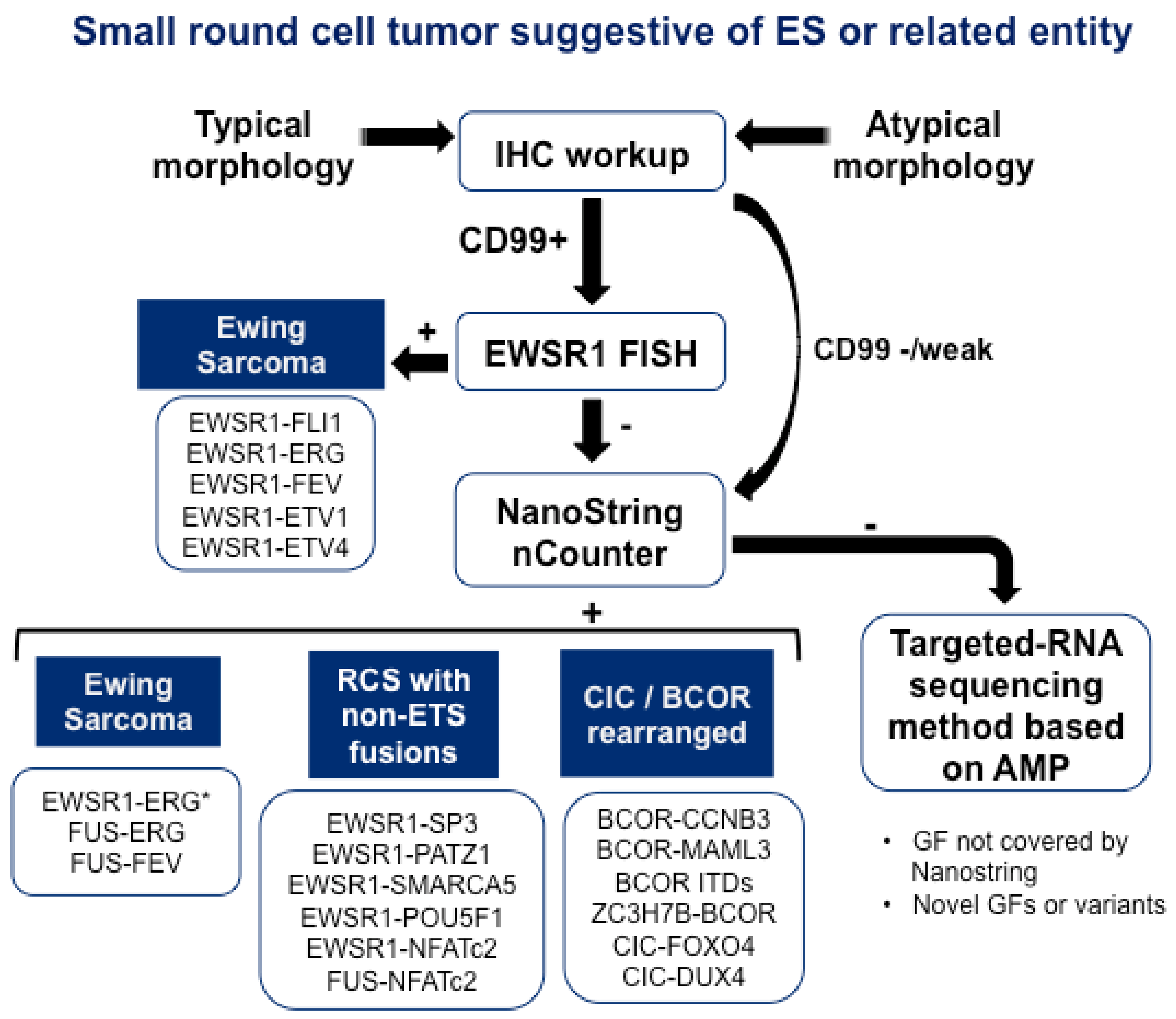
2. Improvement in the Diagnosis and Discovery of New Entities Using Next-Generation Techniques
3. The Heterogeneous Molecular Phenotype of Ewing Sarcoma
4. Advances and Utility of Liquid Biopsy-Based Studies
5. Unveiling New Molecular Targets Based on Pre-Clinical Studies
6. Future Directions on Ewing Sarcoma Research
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMP | Anchored Multiplex PCR |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CTC | Circulating tumor cells |
| ctDNA | Circulating tumor DNA |
| ctRNA | Circulating tumor RNA |
| ddPCR | Droplet digital PCR |
| DSBs | Double Strand Breaks |
| DSRCT | Desmoplastic Small Round Blue Cell Tumors |
| ELS | Ewing-like Sarcoma |
| ES | Ewing Sarcoma |
| EVs | Extracellular Vesicles |
| FISH | Fluorescence In Situ Hybridization |
| HR | Homologous Recombination |
| IHC | Immunohistochemistry |
| ITH | Intratumoral heterogeneity |
| LB | Liquid biopsy |
| MSC | Mesenchymal Stem Cells |
| NGS | Next Generation Sequencing |
| ORF | Open Reading Frames |
| PARPi | PARP inhibitors |
| PDX | Patient Derived Xenografts |
| PET | Positron Emission Tomography |
| RT-PCR | Reverse Transcriptase-PCR |
| shRNA | Short hairpin RNA |
| SNP | Single nucleotide polymorphism |
| SNV | Single-Nucleotide Variants |
| SSBs | Single Strand Breaks |
| WGS | Whole Genome Sequencing |
References
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers. 2018, 4, 5. [Google Scholar] [CrossRef]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.J.; Han, J.; Han, Z.Y.; Sax, B.; Kream, B.E.; et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017, 8, 34141–34163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.C. Organoids: Avatars for Personalized Medicine. Keio J. Med. 2019, 68, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driehuis, E.; Van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Gardner, J.M.; Lucas, D.R.; McHugh, J.B.; Patel, R.M. Ewing sarcoma. Semin. Diagn. Pathol. 2014, 31, 39–47. [Google Scholar] [CrossRef]
- Aurias, A.; Rimbaut, C.; Buffe, D.; Zucker, J.M.; Mazabraud, A. Translocation involving chromosome 22 in Ewing’s sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet. Cytogenet. 1984, 12, 21–25. [Google Scholar] [CrossRef]
- Russell-Goldman, E.; Hornick, J.L.; Qian, X.; Jo, V. NKX2.2 immunohistochemistry in the distinction of Ewing sarcoma from cytomorphologic mimics: Diagnostic utility and pitfalls. Cancer Cytopathol. 2018, 126, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; Yoshida, K.; Kaneko, Y.; Tanaka, Y.; Sasaki, Y.; Urano, F.; Umezawa, A.; Hata, J.; Fujinaga, K. The genomic breakpoint and chimeric transcripts in the EWSR1-ETV4/E1AF GF in Ewing sarcoma. Cytogenet Cell Genet. 1998, 82, 278–283. [Google Scholar] [CrossRef]
- Jeon, I.S.; Davis, J.N.; Braun, B.S.; E Sublett, J.; Roussel, M.F.; Denny, C.T.; Shapiro, D.N. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995, 10, 1229–1234. [Google Scholar] [PubMed]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bhargava, R.; Zheng, T.; Wexler, L.; Collins, M.H.; Roulston, D.; Ladanyi, M. Undifferentiated small round cell sarcomas with rare EWS GFs: identification of a novel EWS-SP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J. Mol. Diagn. 2007, 9, 498–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- . Renzi, S.; Anderson, N.D.; Light, N.; Gupta, A. Ewing-like sarcoma: An emerging family of round cell sarcomas. J. Cell. Physiol. 2018, 234, 7999–8007. [Google Scholar] [CrossRef]
- Sankar, S.; Lessnick, S.L. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-L.; Patel, N.R.; Caragea, M.; Hogendoorn, P.C.W.; López-Terrada, L.; Hornick, J.L.; Lazar, A.J.; L, D. Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod. Pathol. 2012, 25, 1378–1383. [Google Scholar] [CrossRef]
- Charville, G.W.; Wang, W.-L.; Ingram, D.R.; Roy, A.; Thomas, D.; Patel, R.M.; Hornick, J.L.; Van De Rijn, M.; Lazar, A.J. EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod. Pathol. 2017, 30, 1312–1320. [Google Scholar] [CrossRef]
- Carter, C.; Patel, R.M. Important Recently Characterized Non-Ewing Small Round Cell Tumors. Surg. Pathol. Clin. 2018, 12, 191–215. [Google Scholar] [CrossRef]
- Le Loarer, F.; Pissaloux, D.; Coindre, J.M.; Tirode, F.; Vince, D.R. Update on Families of Round Cell Sarcomas Other than Classical Ewing Sarcomas. Surg. Pathol. Clin. 2017, 10, 587–620. [Google Scholar] [CrossRef]
- Chen, S.; Deniz, K.; Sung, Y.S.; Zhang, L.; Dry, S.; Antonescu, C.R. Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 2016, 55, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Noujaim, J.; Jones, R.L.; Swansbury, J.; Gonzalez, D.; Benson, C.; Judson, I.; Fisher, C.; Thway, K. The spectrum of EWSR1-rearranged neoplasms at a tertiary sarcoma centre; assessing 772 tumour specimens and the value of current ancillary molecular diagnostic modalities. Br. J. Cancer 2017, 116, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Brcic, I.; Brodowicz, T.; Cerroni, L.; Kashofer, K.; Serbanescu, G.L.; Kasseroler, M.T.; Amann, G.; Scheipl, S.; Szkandera, J.; Leithner, A.; et al. Undifferentiated round cell sarcomas with CIC-DUX4 GF: expanding the clinical spectrum. Pathology 2020, 52, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krystel-Whittemore, M.; Taylor, M.S.; Rivera, M.; Lennerz, J.K.; Le, L.P.; Dias-Santagata, D.; Iafrate, A.J.; Deshpande, V.; Chebib, I.; Nielsen, G.P.; et al. Novel and established EWSR1 GFs and associations identified by next-generation sequencing and fluorescence in-situ hybridization. Hum. Pathol 2019, 93, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.T.-E.; Goytain, A.; Tucker, T.; Karsan, A.; Lee, C.-H.; Nielsen, T.O.; Ng, T.L. Development and Evaluation of a Pan-Sarcoma Fusion Gene Detection Assay Using the NanoString nCounter Platform. J. Mol. Diagn. 2018, 20, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Sheth, J.; Arnoldo, A.; Zhong, Y.; Marrano, P.; Pereira, C.; Ryall, S.; Thorner, P.; Hawkins, C.; Somers, G.R. Sarcoma Subgrouping by Detection of Fusion Transcripts Using NanoString nCounter Technology. Pediatr. Dev. Pathol. 2018, 22, 205–213. [Google Scholar] [CrossRef]
- Song, W.; Platteel, I.; Suurmeijer, A.J.H.; Van Kempen, L.C. Diagnostic yield of NanoString nCounter FusionPlex profiling in soft tissue tumors. Genes, Chromosom. Cancer 2020, 59, 318–324. [Google Scholar]
- Mackintosh, C.; Ordóñez, J.L.; Garcia, D.J.; Sevillano, V.; Llombart-Bosch, A.; Szuhai, K.; Scotlandi, K.; Alberghini, M.; Sciot, R.; Sinnaeve, F.; et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 2011, 31, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Brohl, A.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The Genomic Landscape of the Ewing Sarcoma Family of Tumors Reveals Recurrent STAG2 Mutation. Plos Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The Genomic Landscape of Pediatric Ewing Sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Huertas-Martínez, J.; Court, F.; Rello-Varona, S.; Herrero-Martín, D.; Almacellas-Rabaiget, O.; Sáinz-Jaspeado, M.; Garcia-Monclús, S.; Lagares-Tena, L.; Buj, R.; Hontecillas-Prieto, L.; et al. DNA methylation profiling identifies PTRF/Cavin-1 as a novel tumor suppressor in Ewing sarcoma when co-expressed with caveolin-1. Cancer Lett. 2017, 386, 196–207. [Google Scholar] [CrossRef]
- Katschnig, A.M.; Kauer, M.O.; Schwentner, R.; Tomazou, E.M.; Mutz, C.N.; Linder, M.; Sibilia, M.; Alonso, J.; Aryee, D.N.T.; Kovar, H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017, 36, 5995–6005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelsche, C.; Kriegsmann, M.; Kommoss, F.K.F.; Stichel, D.; Kriegsmann, K.; Vokuhl, C.; Grünewald, T.G.P.; Romero-Pérez, L.; Kirchner, T.; De Alava, E.; et al. DNA methylation profiling distinguishes Ewing-like sarcoma with EWSR1–NFATc2 fusion from Ewing sarcoma. J. Cancer Res. Clin. Oncol. 2019, 145, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Romero-Pérez, L.; Surdez, D.; Brunet, E.; Delattre, O.; Grünewald, T.G.P. STAG Mutations in Cancer. Trends Cancer 2019, 5, 506–520. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.D.; Le Deley, M.-C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetête-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.D.; de Borja, R.; Young, M.D.; Fuligni, F.; Rosic, A.; Roberts, N.D.; Hajjar, S.; Layeghifard, M.; Novokmet, A.; Kowalski, P.E.; et al. Rearrangement bursts generate canonical GFs in bone and soft tissue tumors. Science 2018, 361. [Google Scholar]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; Macdonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Franzetti, G.-A.; Laud-Duval, K.; Van Der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; De Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Krumbholz, M.; Hellberg, J.; Steif, B.; Bäuerle, T.; Gillmann, C.; Fritscher, T.; Agaimy, A.; Frey, B.; Juengert, J.; Wardelmann, E.; et al. Genomic EWSR1 Fusion Sequence as Highly Sensitive and Dynamic Plasma Tumor Marker in Ewing Sarcoma. Clin. Cancer Res. 2016, 22, 4356–4365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, N.N.; Patel, J.A.; Magnan, H.; Zehir, A.; You, D.; Tang, J.; Meng, F.; Samoila, A.; Slotkin, E.K.; Ambati, S.R.; et al. Plasma DNA-Based Molecular Diagnosis, Prognostication, and Monitoring of Patients With EWSR1 Fusion-Positive Sarcomas. JCO Precis. Oncol. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Shulman, D.S.; Klega, K.; Imamovic-Tuco, A.; Clapp, A.; Nag, A.; Thorner, A.R.; Van Allen, E.; Ha, G.; Lessnick, S.L.; Gorlick, R.; et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group. Br. J. Cancer 2018, 119, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Casini, B.; Mandoj, C.; Benini, S.; Alberti, L.; Novello, M.; Melucci, E.; Conti, L.; Covello, R.; Pescarmona, E.; et al. Precision diagnostics of Ewing’s sarcoma by liquid biopsy: circulating EWS-FLI1 fusion transcripts. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, I.V.; Raposo, G.; Welsch, U.; Da Costa, O.P.; Thiel, U.; Lebar, M.; Maurer, M.; Bender, H.-U.; Von Luettichau, I.; Richter, G.H.S.; et al. First identification of Ewing’s sarcoma-derived extracellular vesicles and exploration of their biological and potential diagnostic implications. Boil. Cell 2013, 105, 289–303. [Google Scholar] [CrossRef]
- Evdokimova, V.; Ruzanov, P.; Gassmann, H.; Zaidi, S.H.; Peltekova, V.; E Heisler, L.; McPherson, J.D.; Orlic-Milacic, M.; Specht, K.; Steiger, K.; et al. Exosomes transmit retroelement RNAs to drive inflammation and immunosuppression in Ewing Sarcoma. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- A May, W.; Lessnick, S.L.; Braun, B.S.; Klemsz, M.; Lewis, B.C.; Lunsford, L.B.; Hromas, R.; Denny, C.T. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol. Cell. Boil. 1993, 13, 7393–7398. [Google Scholar] [CrossRef] [Green Version]
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Research 2019, 8, 493. [Google Scholar] [CrossRef] [Green Version]
- Grohar, P.J.; Kim, S.; Rivera, G.O.R.; Sen, N.; Haddock, S.; Harlow, M.L.; Maloney, N.K.; Zhu, J.; O’Neill, M.; Jones, T.L.; et al. Functional Genomic Screening Reveals Splicing of the EWS-FLI1 Fusion Transcript as a Vulnerability in Ewing Sarcoma. Cell Rep. 2016, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Harlow, M.L.; Maloney, N.; Roland, J.; Navarro, M.J.G.; Easton, M.K.; Kitchen-Goosen, S.M.; Boguslawski, E.A.; Madaj, Z.B.; Johnson, B.K.; Bowman, M.; et al. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016, 76, 6657–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Sankhala, K.K.; Ratan, R.; Garcia, E.S.; Boni, V.; Gil, T.; Villalobos, V.M.; Chawla, S.P.; Lardelli, P.; Siguero, M.; et al. Efficacy and safety of lurbinectedin (PM1183) in Ewing sarcoma: Final results from a phase 2 study. J. Clin. Oncol 2018, 36, 39. [Google Scholar] [CrossRef]
- Fidaleo, M.; De Paola, E.; Paronetto, M.P. The RNA helicase A in malignant transformation. Oncotarget 2016, 7, 28711–28723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.-H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Boil. 2017, 18, 610–621. [Google Scholar]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS–FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Choy, E.; E Butrynski, J.; Harmon, D.C.; A Morgan, J.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Yoon, C.; Schmidt, B.; Park, J.; Zhang, A.Y.; Erkizan, H.V.; Toretsky, J.A.; Kirsch, D.G.; Lin, J.-X. Combining PARP-1 inhibition and radiation in Ewing sarcoma results in lethal DNA damage. Mol. Cancer Ther. 2013, 12, 2591–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.A.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Soler, M.; Morgado-Palacin, I.; Lafarga, V.; Lecona, E.; Murga, M.; Callen, E.; Azorin, D.; Alonso, J.; Lopez-Contreras, A.J.; Nussenzweig, A.; et al. Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget 2016, 7, 58759–58767. [Google Scholar] [CrossRef] [Green Version]
- Countryman, P.; Fan, Y.; Gorthi, A.; Pan, H.; Strickland, J.; Kaur, P.; Wang, X.; Lin, J.; Lei, X.; White, C.; et al. Cohesin SA2 is a sequence-independent DNA-binding protein that recognizes DNA replication and repair intermediates. J. Boil. Chem. 2017, 293, 1054–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, G.; Stevers, M.; Goode, B.; Ashworth, A.; Solomon, D.A. A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat. Commun. 2019, 10, 1686. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef] [Green Version]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L., 2nd; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res 2019, 25, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iniguez, A.B.; Stolte, B.; Wang, E.J.; Conway, A.S.; Alexe, G.; Dharia, N.V.; Kwiatkowski, N.; Zhang, T.; Abraham, B.J.; Mora, J.; et al. EWS/FLI Confers Tumor Cell Synthetic Lethality to CDK12 Inhibition in Ewing Sarcoma. Cancer Cell 2018, 33, 202–216.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neilsen, P.; Pishas, K.I.; Callen, D.; Thomas, D.M. Targeting the p53 Pathway in Ewing Sarcoma. Sarcoma 2010, 2011, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Stolte, B.; Iniguez, A.B.; Dharia, N.V.; Robichaud, A.L.; Conway, A.S.; Morgan, A.M.; Alexe, G.; Schauer, N.J.; Liu, X.; Bird, G.H.; et al. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Johnson, K.; Taslim, C.; Saund, R.S.; Lessnick, S.L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLoS ONE 2017, 12, e0186275. [Google Scholar] [CrossRef]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G.P. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef]
- Musa, J.; Cidre-Aranaz, F.; Aynaud, M.-M.; Orth, M.F.; Knott, M.M.L.; Mirabeau, O.; Mazor, G.; Varon, M.; Hölting, T.L.B.; Grossetête, S.; et al. Cooperation of cancer drivers with regulatory germline variants shapes clinical outcomes. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Avcı, Ç.B. Monoclonal antibodies in cancer immunotherapy. Mol. Boil. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef]
- Puerto-Camacho, P.; Amaral, A.T.; Lamhamedi-Cherradi, S.-E.; Menegaz, B.A.; Castillo-Ecija, H.; Ordóñez, J.L.; Domínguez, S.; Jordan-Perez, C.; Díaz-Martín, J.; Romero-Pérez, L.; et al. Preclinical Efficacy of Endoglin-Targeting Antibody–Drug Conjugates for the Treatment of Ewing Sarcoma. Clin. Cancer Res. 2018, 25, 2228–2240. [Google Scholar] [CrossRef]
- Thiel, U.; Schober, S.J.; Einspieler, I.; Kirschner, A.; Thiede, M.; Schirmer, D.; Gall, K.; Blaeschke, F.; Schmidt, O.; Jabar, S.; et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. OncoImmunology 2017, 6, e1312239. [Google Scholar] [CrossRef] [PubMed]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019, 15, 100221. [Google Scholar] [CrossRef] [PubMed]
- Aynaud, M.-M.; Mirabeau, O.; Gruel, N.; Grossetête, S.; Boeva, V.; Durand, S.; Surdez, D.; Saulnier, O.; Zaïdi, S.; Gribkova, S.; et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 2020, 30, 1767–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salguero-Aranda, C.; Amaral, A.T.; Olmedo-Pelayo, J.; Diaz-Martin, J.; Álava, E.d. Breakthrough Technologies Reshape the Ewing Sarcoma Molecular Landscape. Cells 2020, 9, 804. https://doi.org/10.3390/cells9040804
Salguero-Aranda C, Amaral AT, Olmedo-Pelayo J, Diaz-Martin J, Álava Ed. Breakthrough Technologies Reshape the Ewing Sarcoma Molecular Landscape. Cells. 2020; 9(4):804. https://doi.org/10.3390/cells9040804
Chicago/Turabian StyleSalguero-Aranda, Carmen, Ana Teresa Amaral, Joaquín Olmedo-Pelayo, Juan Diaz-Martin, and Enrique de Álava. 2020. "Breakthrough Technologies Reshape the Ewing Sarcoma Molecular Landscape" Cells 9, no. 4: 804. https://doi.org/10.3390/cells9040804
APA StyleSalguero-Aranda, C., Amaral, A. T., Olmedo-Pelayo, J., Diaz-Martin, J., & Álava, E. d. (2020). Breakthrough Technologies Reshape the Ewing Sarcoma Molecular Landscape. Cells, 9(4), 804. https://doi.org/10.3390/cells9040804