The Cuprizone Model: Dos and Do Nots

 , and
, and 
Abstract
:1. Introduction
2. Characteristics of the Cuprizone Model
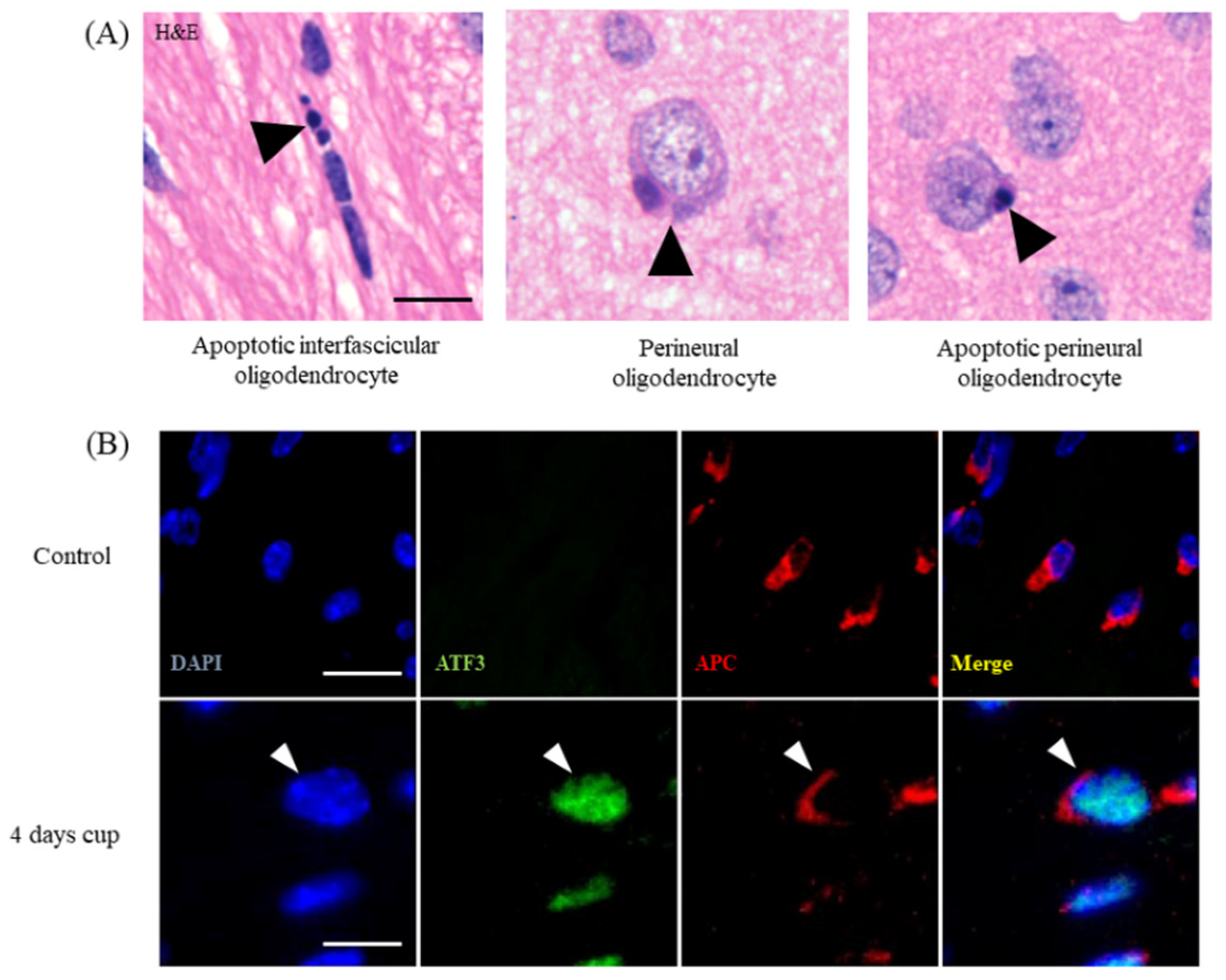
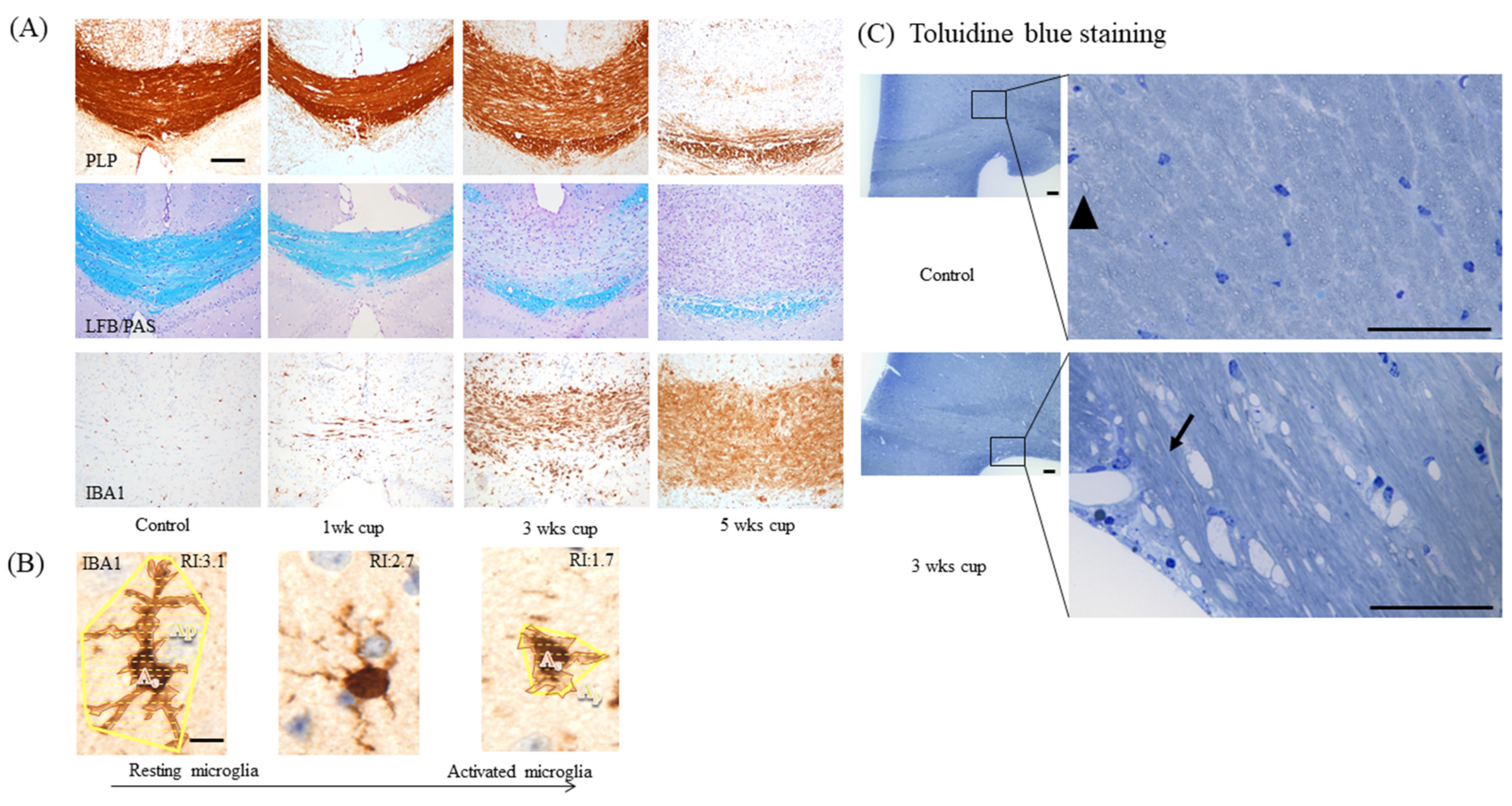
2.1. Week 1
2.2. Weeks 1–3
2.3. Weeks 3–5
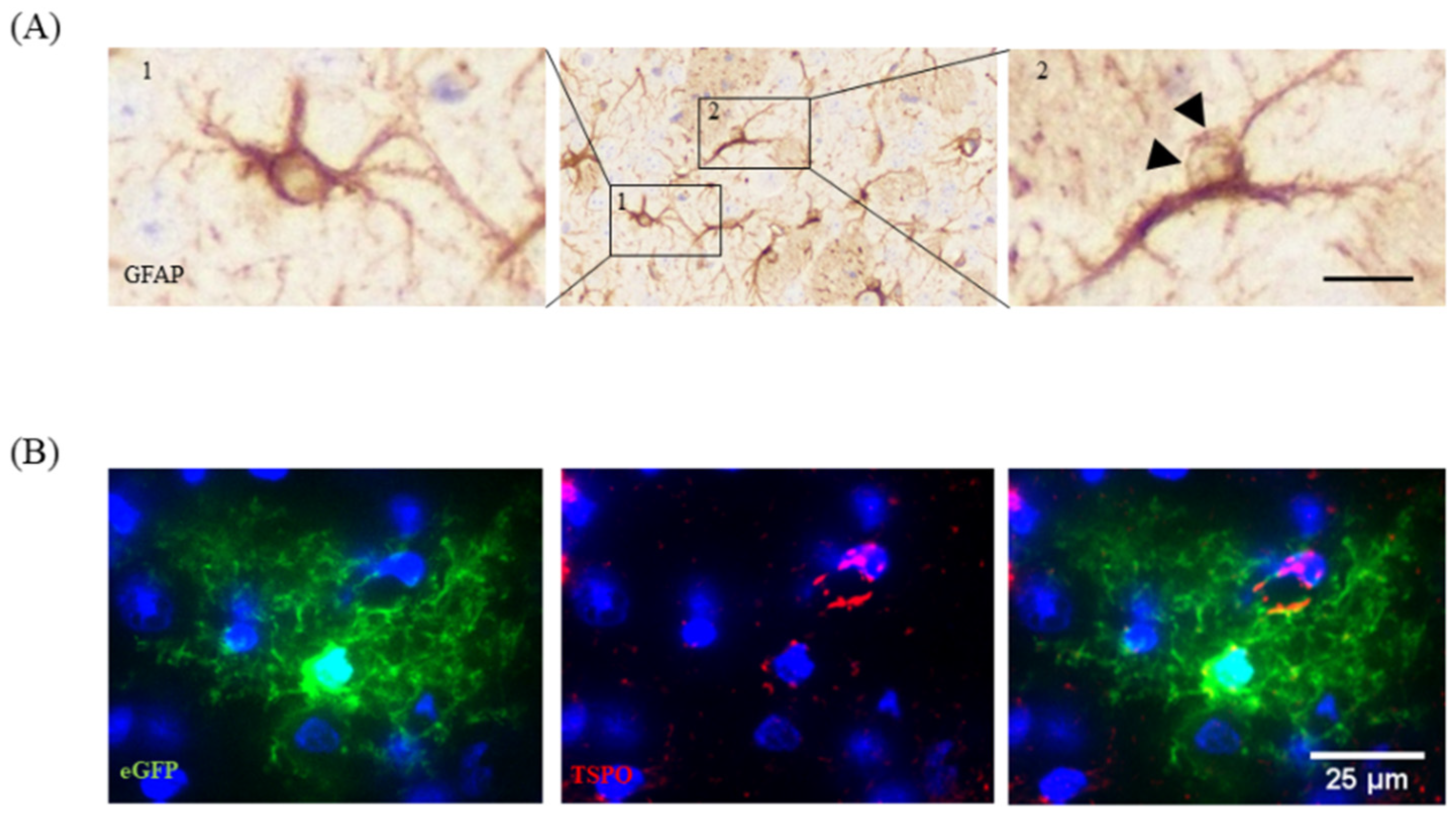
3. Dos and Do Nots in the Cuprizone Model
3.1. Animal Weight
3.2. Cuprizone Formulation
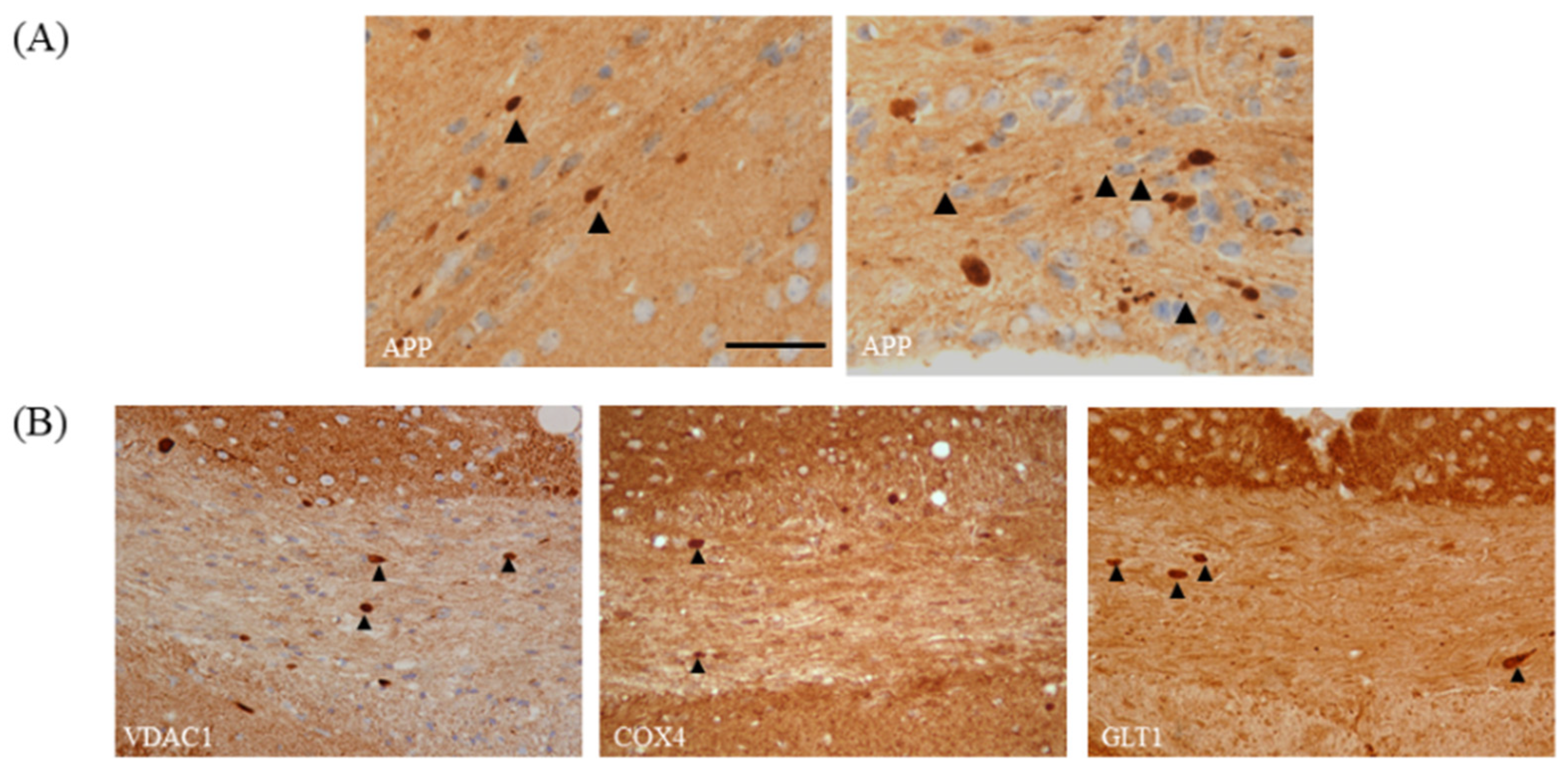
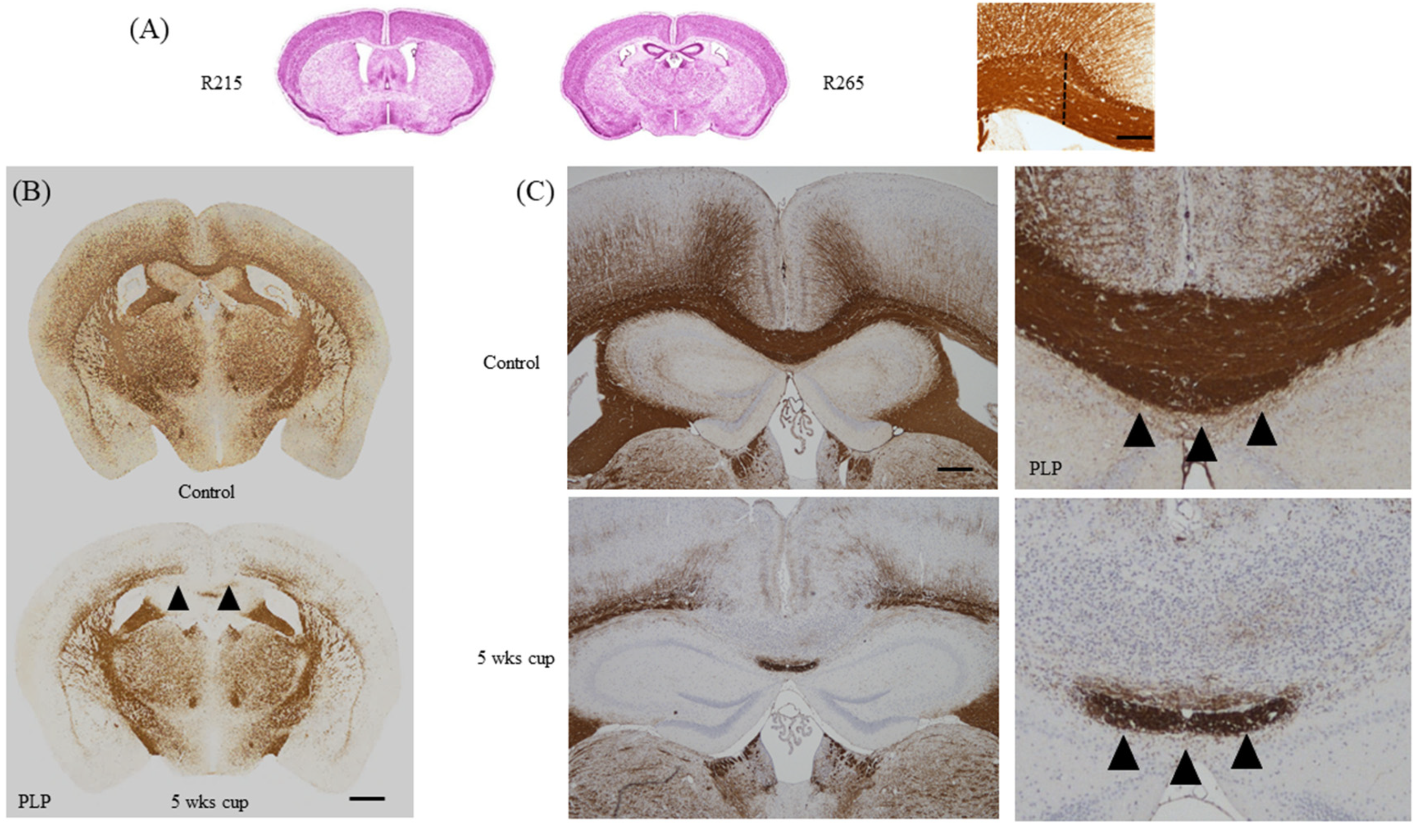
3.3. Selection of the Region of Interest for Histological Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carassiti, D.; Altmann, D.R.; Petrova, N.; Pakkenberg, B.; Scaravilli, F.; Schmierer, K. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol. Appl. Neurobiol. 2018, 44, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Chang, A.; Doud, M.K.; Kidd, G.J.; Ribaudo, M.V.; Young, E.A.; Fox, R.J.; Staugaitis, S.M.; Trapp, B.D. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011, 69, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, T.; Jafari, M.; Kreutzfeldt, M.; Bahn, E.; Bruck, W.; Kerschensteiner, M.; Merkler, D. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 2016, 139, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. New Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W.; Bo, L.; Mork, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef]
- Naldi, P.; Collimedaglia, L.; Vecchio, D.; Rosso, M.G.; Perl, F.; Stecco, A.; Monaco, F.; Leone, M.A. Predictors of attack severity and duration in multiple sclerosis: A prospective study. Open Neurol. J. 2011, 5, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Siffrin, V.; Radbruch, H.; Glumm, R.; Niesner, R.; Paterka, M.; Herz, J.; Leuenberger, T.; Lehmann, S.M.; Luenstedt, S.; Rinnenthal, J.L.; et al. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity 2010, 33, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Aktas, O.; Smorodchenko, A.; Brocke, S.; Infante-Duarte, C.; Schulze Topphoff, U.; Vogt, J.; Prozorovski, T.; Meier, S.; Osmanova, V.; Pohl, E.; et al. Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 2005, 46, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.J. Sodium channels and multiple sclerosis: Roles in symptom production, damage and therapy. Brain Pathol. 2007, 17, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.L.; Garg, P.; Thin, T.H.; Yoo, S.; Dutta, R.; Trapp, B.D.; Haroutunian, V.; Zhu, J.; Donovan, M.J.; Sharp, A.J.; et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci. 2014, 17, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Elkjaer, M.L.; Frisch, T.; Reynolds, R.; Kacprowski, T.; Burton, M.; Kruse, T.A.; Thomassen, M.; Baumbach, J.; Illes, Z. Unique RNA signature of different lesion types in the brain white matter in progressive multiple sclerosis. Acta Neuropathol. Commun. 2019, 7, 58. [Google Scholar] [CrossRef]
- Behrangi, N.; Fischbach, F.; Kipp, M. Mechanism of Siponimod: Anti-Inflammatory and Neuroprotective Mode of Action. Cells 2019, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Tremlett, H.; Zhao, Y.; Joseph, J.; Devonshire, V. Relapses in multiple sclerosis are age- and time-dependent. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1368–1374. [Google Scholar] [CrossRef]
- Bolton, C.; Smith, P.A. The influence and impact of ageing and immunosenescence (ISC) on adaptive immunity during multiple sclerosis (MS) and the animal counterpart experimental autoimmune encephalomyelitis (EAE). Ageing Res. Rev. 2018, 41, 64–81. [Google Scholar] [CrossRef]
- Penner, I.K.; Aktas, O. Functional reorganization is a maladaptive response to injury—NO. Mult. Scler. 2017, 23, 193–194. [Google Scholar] [CrossRef] [Green Version]
- Enzinger, C.; Pinter, D.; Rocca, M.A.; De Luca, J.; Sastre-Garriga, J.; Audoin, B.; Filippi, M. Longitudinal fMRI studies: Exploring brain plasticity and repair in MS. Mult. Scler. 2016, 22, 269–278. [Google Scholar] [CrossRef]
- Kipp, M.; Clarner, T.; Dang, J.; Copray, S.; Beyer, C. The cuprizone animal model: New insights into an old story. Acta Neuropathol. 2009, 118, 723–736. [Google Scholar] [CrossRef]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, S.A.; Duking, T.; Spieth, L.; Winchenbach, J.; Stumpf, S.K.; Gerndt, N.; Kusch, K.; Ruhwedel, T.; Mobius, W.; Saher, G. Blood-brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol. Commun. 2017, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.; Shemer, A.; Levy-Efrati, L.; Gross, M.; Kim, J.S.; Engel, A.; David, E.; Chappell-Maor, L.; Grozovski, J.; Rotkopf, R.; et al. Microglial MHC class II is dispensable for experimental autoimmune encephalomyelitis and cuprizone-induced demyelination. Eur. J. Immunol. 2018, 48, 1308–1318. [Google Scholar] [CrossRef]
- Hiremath, M.M.; Chen, V.S.; Suzuki, K.; Ting, J.P.; Matsushima, G.K. MHC class II exacerbates demyelination in vivo independently of T cells. J. Neuroimmunol. 2008, 203, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slowik, A.; Schmidt, T.; Beyer, C.; Amor, S.; Clarner, T.; Kipp, M. The sphingosine 1-phosphate receptor agonist FTY720 is neuroprotective after cuprizone-induced CNS demyelination. Br. J. Pharmacol. 2015, 172, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voskuhl, R.R.; Itoh, N.; Tassoni, A.; Matsukawa, M.A.; Ren, E.; Tse, V.; Jang, E.; Suen, T.T.; Itoh, Y. Gene expression in oligodendrocytes during remyelination reveals cholesterol homeostasis as a therapeutic target in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 10130–10139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, M.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci. Lett. 2009, 453, 120–125. [Google Scholar] [CrossRef]
- Zendedel, A.; Beyer, C.; Kipp, M. Cuprizone-induced demyelination as a tool to study remyelination and axonal protection. J. Mol. Neurosci. 2013, 51, 567–572. [Google Scholar] [CrossRef]
- Kipp, M. Remyelination strategies in multiple sclerosis: A critical reflection. Expert Rev. Neurother. 2016, 16, 1–3. [Google Scholar] [CrossRef]
- Shields, S.A.; Gilson, J.M.; Blakemore, W.F.; Franklin, R.J. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia 1999, 28, 77–83. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baror, R.; Neumann, B.; Segel, M.; Chalut, K.J.; Fancy, S.P.J.; Schafer, D.P.; Franklin, R.J.M. Transforming growth factor-beta renders ageing microglia inhibitory to oligodendrocyte generation by CNS progenitors. Glia 2019, 67, 1374–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beissel, C.; van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage-inducible transcript 3. Glia 2019, 67, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Krauspe, B.M.; Dreher, W.; Beyer, C.; Baumgartner, W.; Denecke, B.; Janssen, K.; Langhans, C.D.; Clarner, T.; Kipp, M. Short-term cuprizone feeding verifies N-acetylaspartate quantification as a marker of neurodegeneration. J. Mol. Neurosci. 2015, 55, 733–748. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Kim, W.K.; Kim, D.; Cui, J.; Jang, H.H.; Kim, K.S.; Lee, H.J.; Kim, S.U.; Ahn, S.M. Secretome analysis of human oligodendrocytes derived from neural stem cells. PLoS ONE 2014, 9, e84292. [Google Scholar] [CrossRef] [Green Version]
- Kipp, M.; Gingele, S.; Pott, F.; Clarner, T.; van der Valk, P.; Denecke, B.; Gan, L.; Siffrin, V.; Zipp, F.; Dreher, W.; et al. BLBP-expression in astrocytes during experimental demyelination and in human multiple sclerosis lesions. Brain Behav. Immun. 2011, 25, 1554–1568. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Boeda, B.; Etienne-Manneville, S. APC binds intermediate filaments and is required for their reorganization during cell migration. J. Cell Biol. 2013, 200, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.N.; Jeon, G.S.; Kim, D.W.; Cho, I.H.; Cho, S.S. Expression of adenomatous polyposis coli protein in reactive astrocytes in hippocampus of kainic acid-induced rat. Neurochem. Res. 2010, 35, 114–121. [Google Scholar] [CrossRef]
- Salinas Tejedor, L.; Gudi, V.; Kucman, V.; Pul, R.; Gingele, S.; Suhs, K.W.; Stangel, M.; Skripuletz, T. Oligodendroglial markers in the cuprizone model of CNS de- and remyelination. Histol. Histopathol. 2015, 30, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, J.P.; Berger, K.; Awad, H.; Clarner, T.; Beyer, C.; Kipp, M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J. Mol. Neurosci. 2012, 48, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced de- and remyelination in the CNS: Lessons learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Nack, A.; Brendel, M.; Nedelcu, J.; Daerr, M.; Nyamoya, S.; Beyer, C.; Focke, C.; Deussing, M.; Hoornaert, C.; Ponsaerts, P.; et al. Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells 2019, 8, 94. [Google Scholar] [CrossRef] [Green Version]
- Nolte, C.; Matyash, M.; Pivneva, T.; Schipke, C.G.; Ohlemeyer, C.; Hanisch, U.K.; Kirchhoff, F.; Kettenmann, H. GFAP promoter-controlled EGFP-expressing transgenic mice: A tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 2001, 33, 72–86. [Google Scholar] [CrossRef]
- Chrzanowski, U.; Schmitz, C.; Horn-Bochtler, A.; Nack, A.; Kipp, M. Evaluation strategy to determine reliable demyelination in the cuprizone model. Metab. Brain Dis. 2019, 34, 681–685. [Google Scholar] [CrossRef]
- Grosse-Veldmann, R.; Becker, B.; Amor, S.; van der Valk, P.; Beyer, C.; Kipp, M. Lesion Expansion in Experimental Demyelination Animal Models and Multiple Sclerosis Lesions. Mol. Neurobiol. 2016, 53, 4905–4917. [Google Scholar] [CrossRef] [PubMed]
- Clarner, T.; Diederichs, F.; Berger, K.; Denecke, B.; Gan, L.; van der Valk, P.; Beyer, C.; Amor, S.; Kipp, M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 2012, 60, 1468–1480. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Sisodia, S.S.; Archer, D.R.; Martin, L.J.; Weidemann, A.; Beyreuther, K.; Fischer, P.; Masters, C.L.; Price, D.L. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. USA 1990, 87, 1561–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherriff, F.E.; Bridges, L.R.; Gentleman, S.M.; Sivaloganathan, S.; Wilson, S. Markers of axonal injury in post mortem human brain. Acta Neuropathol. 1994, 88, 433–439. [Google Scholar] [CrossRef]
- Stone, J.R.; Singleton, R.H.; Povlishock, J.T. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): A site specific marker for the detection of traumatic axonal injury. Brain Res. 2000, 871, 288–302. [Google Scholar] [CrossRef]
- Hoflich, K.M.; Beyer, C.; Clarner, T.; Schmitz, C.; Nyamoya, S.; Kipp, M.; Hochstrasser, T. Acute axonal damage in three different murine models of multiple sclerosis: A comparative approach. Brain Res. 2016, 1650, 125–133. [Google Scholar] [CrossRef]
- Ruhling, S.; Kramer, F.; Schmutz, S.; Amor, S.; Jiangshan, Z.; Schmitz, C.; Kipp, M.; Hochstrasser, T. Visualization of the Breakdown of the Axonal Transport Machinery: A Comparative Ultrastructural and Immunohistochemical Approach. Mol. Neurobiol. 2019, 56, 3984–3998. [Google Scholar] [CrossRef]
- Thiessen, J.D.; Zhang, Y.; Zhang, H.; Wang, L.; Buist, R.; Del Bigio, M.R.; Kong, J.; Li, X.M.; Martin, M. Quantitative MRI and ultrastructural examination of the cuprizone mouse model of demyelination. NMR Biomed. 2013, 26, 1562–1581. [Google Scholar] [CrossRef]
- Acs, P.; Komoly, S. Selective ultrastructural vulnerability in the cuprizone-induced experimental demyelination. Ideggyogy. Szle. 2012, 65, 266–270. [Google Scholar]
- Wergeland, S.; Torkildsen, O.; Myhr, K.M.; Mork, S.J.; Bo, L. The cuprizone model: Regional heterogeneity of pathology. APMIS 2012, 120, 648–657. [Google Scholar] [CrossRef]
- Torkildsen, O.; Brunborg, L.A.; Myhr, K.M.; Bo, L. The cuprizone model for demyelination. Acta Neurol. Scand. 2008, 188, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Mahamid, J. Unravelling molecular complexity in structural cell biology. Curr. Opin. Struct. Biol. 2018, 52, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Brousse, B.; Magalon, K.; Durbec, P.; Cayre, M. Region and dynamic specificities of adult neural stem cells and oligodendrocyte precursors in myelin regeneration in the mouse brain. Biol. Open 2015, 4, 980–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Yang, H.J.; McConomy, B.; Browning, R.; Li, X.M. Behavioral and neurobiological changes in C57BL/6 mouse exposed to cuprizone: Effects of antipsychotics. Front. Behav. Neurosci. 2010, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Hibbits, N.; Pannu, R.; Wu, T.J.; Armstrong, R.C. Cuprizone demyelination of the corpus callosum in mice correlates with altered social interaction and impaired bilateral sensorimotor coordination. Asn Neuro 2009, 1. [Google Scholar] [CrossRef]
- Liebetanz, D.; Merkler, D. Effects of commissural de- and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp. Neurol. 2006, 202, 217–224. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Boretius, S.; Ott, C.; Von Streitberg, A.; Welpinghus, H.; Sperling, S.; Frahm, J.; Simons, M.; Ghezzi, P.; Ehrenreich, H. Erythropoietin attenuates neurological and histological consequences of toxic demyelination in mice. Mol. Med. 2012, 18, 628–635. [Google Scholar] [CrossRef]
- Manrique-Hoyos, N.; Jurgens, T.; Gronborg, M.; Kreutzfeldt, M.; Schedensack, M.; Kuhlmann, T.; Schrick, C.; Bruck, W.; Urlaub, H.; Simons, M.; et al. Late motor decline after accomplished remyelination: Impact for progressive multiple sclerosis. Ann. Neurol. 2012, 71, 227–244. [Google Scholar] [CrossRef]
- Franco-Pons, N.; Torrente, M.; Colomina, M.T.; Vilella, E. Behavioral deficits in the cuprizone-induced murine model of demyelination/remyelination. Toxicol. Lett. 2007, 169, 205–213. [Google Scholar] [CrossRef]
- Zhan, J.; Yakimov, V.; Ruhling, S.; Fischbach, F.; Nikolova, E.; Joost, S.; Kaddatz, H.; Greiner, T.; Frenz, J.; Holzmann, C.; et al. High Speed Ventral Plane Videography as a Convenient Tool to Quantify Motor Deficits during Pre-Clinical Experimental Autoimmune Encephalomyelitis. Cells 2019, 8, 1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinodan, M.; Yamauchi, T.; Tatsumi, K.; Okuda, H.; Takeda, T.; Kiuchi, K.; Sadamatsu, M.; Wanaka, A.; Kishimoto, T. Demyelination in the juvenile period, but not in adulthood, leads to long-lasting cognitive impairment and deficient social interaction in mice. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2009, 33, 978–985. [Google Scholar] [CrossRef]
- Xu, H.; Yang, H.J.; Zhang, Y.; Clough, R.; Browning, R.; Li, X.M. Behavioral and neurobiological changes in C57BL/6 mice exposed to cuprizone. Behav. Neurosci. 2009, 123, 418–429. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharm. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Bruck, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Wagenknecht, N.; Becker, B.; Scheld, M.; Beyer, C.; Clarner, T.; Hochstrasser, T.; Kipp, M. Thalamus Degeneration and Inflammation in Two Distinct Multiple Sclerosis Animal Models. J. Mol. Neurosci. 2016, 60, 102–114. [Google Scholar] [CrossRef]
- Goldberg, J.; Clarner, T.; Beyer, C.; Kipp, M. Anatomical Distribution of Cuprizone-Induced Lesions in C57BL6 Mice. J. Mol. Neurosci. 2015, 57, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Vrenken, H.; Seewann, A.; Knol, D.L.; Polman, C.H.; Barkhof, F.; Geurts, J.J. Diffusely abnormal white matter in progressive multiple sclerosis: In vivo quantitative MR imaging characterization and comparison between disease types. Ajnr Am. J. Neuroradiol. 2010, 31, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Awad, H.; Slowik, A.; Beyer, C.; Kipp, M.; Clarner, T. Regional heterogeneity of cuprizone-induced demyelination: Topographical aspects of the midline of the corpus callosum. J. Mol. Neurosci. 2013, 49, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dallenga, T.; Winkler, A.; Roemer, S.; Maruschak, B.; Siebert, H.; Bruck, W.; Stadelmann, C. Relationship of acute axonal damage, Wallerian degeneration, and clinical disability in multiple sclerosis. J. Neuroinflamm. 2017, 14, 57. [Google Scholar] [CrossRef] [Green Version]
- Haines, J.D.; Inglese, M.; Casaccia, P. Axonal damage in multiple sclerosis. Mt. Sinai J. Med. 2011, 78, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mews, I.; Bergmann, M.; Bunkowski, S.; Gullotta, F.; Bruck, W. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult. Scler. 1998, 4, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Ohno, N.; Chiang, H.; Mahad, D.J.; Kidd, G.J.; Liu, L.; Ransohoff, R.M.; Sheng, Z.H.; Komuro, H.; Trapp, B.D. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc. Natl. Acad. Sci. USA 2014, 111, 9953–9958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado-Santos, J.; Saji, E.; Troscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018, 141, 2066–2082. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Cerina, M.; Narayanan, V.; Gobel, K.; Bittner, S.; Ruck, T.; Meuth, P.; Herrmann, A.M.; Stangel, M.; Gudi, V.; Skripuletz, T.; et al. The quality of cortical network function recovery depends on localization and degree of axonal demyelination. Brain Behav. Immun. 2017, 59, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Remington, L.T.; Babcock, A.A.; Zehntner, S.P.; Owens, T. Microglial recruitment, activation, and proliferation in response to primary demyelination. Am. J. Pathol. 2007, 170, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef]
- Leech, S.; Kirk, J.; Plumb, J.; McQuaid, S. Persistent endothelial abnormalities and blood-brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol. Appl. Neurobiol. 2007, 33, 86–98. [Google Scholar] [CrossRef]
- Plumb, J.; McQuaid, S.; Mirakhur, M.; Kirk, J. Abnormal endothelial tight junctions in active lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. 2002, 12, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: Therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Kashani, I.R.; Chavoshi, H.; Pasbakhsh, P.; Hassani, M.; Omidi, A.; Mahmoudi, R.; Beyer, C.; Zendedel, A. Protective effects of erythropoietin against cuprizone-induced oxidative stress and demyelination in the mouse corpus callosum. Iran. J. Basic Med. Sci. 2017, 20, 886–893. [Google Scholar] [CrossRef]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weissflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Valeiras, B.; Rosato Siri, M.V.; Codagnone, M.; Reines, A.; Pasquini, J.M. Gender influence on schizophrenia-relevant abnormalities in a cuprizone demyelination model. Glia 2014, 62, 1629–1644. [Google Scholar] [CrossRef]
- Taylor, L.C.; Gilmore, W.; Matsushima, G.K. SJL mice exposed to cuprizone intoxication reveal strain and gender pattern differences in demyelination. Brain Pathol. 2009, 19, 467–479. [Google Scholar] [CrossRef]
- Irvine, K.A.; Blakemore, W.F. Age increases axon loss associated with primary demyelination in cuprizone-induced demyelination in C57BL/6 mice. J. Neuroimmunol. 2006, 175, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Leopold, P.; Schmitz, C.; Kipp, M. Animal Weight Is an Important Variable for Reliable Cuprizone-Induced Demyelination. J. Mol. Neurosci. 2019, 68, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Steelman, A.J.; Thompson, J.P.; Li, J. Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci. Res. 2012, 72, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Carey, E.M.; Freeman, N.M. Biochemical changes in Cuprizone-induced spongiform encephalopathy. I. Changes in the activities of 2′,3′-cyclic nucleotide 3′-phosphohydrolase, oligodendroglial ceramide galactosyl transferase, and the hydrolysis of the alkenyl group of alkenyl, acyl-glycerophospholipids by plasmalogenase in different regions of the brain. Neurochem. Res. 1983, 8, 1029–1044. [Google Scholar]
- Soundarapandian, M.M.; Selvaraj, V.; Lo, U.G.; Golub, M.S.; Feldman, D.H.; Pleasure, D.E.; Deng, W. Zfp488 promotes oligodendrocyte differentiation of neural progenitor cells in adult mice after demyelination. Sci. Rep. 2011, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Zatta, P.; Raso, M.; Zambenedetti, P.; Wittkowski, W.; Messori, L.; Piccioli, F.; Mauri, P.L.; Beltramini, M. Copper and zinc dismetabolism in the mouse brain upon chronic cuprizone treatment. Cell. Mol. Life Sci. CMLS 2005, 62, 1502–1513. [Google Scholar] [CrossRef]
- Hochstrasser, T.; Exner, G.L.; Nyamoya, S.; Schmitz, C.; Kipp, M. Cuprizone-Containing Pellets Are Less Potent to Induce Consistent Demyelination in the Corpus Callosum of C57BL/6 Mice. J. Mol. Neurosci. 2017, 61, 617–624. [Google Scholar] [CrossRef]
- Heckers, S.; Held, N.; Kronenberg, J.; Skripuletz, T.; Bleich, A.; Gudi, V.; Stangel, M. Investigation of Cuprizone Inactivation by Temperature. Neurotox. Res. 2017. [Google Scholar] [CrossRef]
- Skripuletz, T.; Lindner, M.; Kotsiari, A.; Garde, N.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am. J. Pathol. 2008, 172, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Herder, V.; Hansmann, F.; Stangel, M.; Skripuletz, T.; Baumgartner, W.; Beineke, A. Lack of cuprizone-induced demyelination in the murine spinal cord despite oligodendroglial alterations substantiates the concept of site-specific susceptibilities of the central nervous system. Neuropathol. Appl. Neurobiol. 2011, 37, 676–684. [Google Scholar] [CrossRef]
- Skripuletz, T.; Bussmann, J.H.; Gudi, V.; Koutsoudaki, P.N.; Pul, R.; Moharregh-Khiabani, D.; Lindner, M.; Stangel, M. Cerebellar cortical demyelination in the murine cuprizone model. Brain Pathol. 2010, 20, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Groebe, A.; Clarner, T.; Baumgartner, W.; Dang, J.; Beyer, C.; Kipp, M. Cuprizone treatment induces distinct demyelination, astrocytosis, and microglia cell invasion or proliferation in the mouse cerebellum. Cerebellum 2009, 8, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Allen Mouse Brain Atlas. Available online: https://mouse.brain-map.org/static/atlas (accessed on 31 March 2020).
- High Resolution Mouse Brain Atlas. Available online: http://www.hms.harvard.edu/research/brain/atlas.html (accessed on 31 March 2020).
- Ruther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Scheld, M.; Ruther, B.J.; Grosse-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymuller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration Triggers Peripheral Immune Cell Recruitment into the Forebrain. J. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
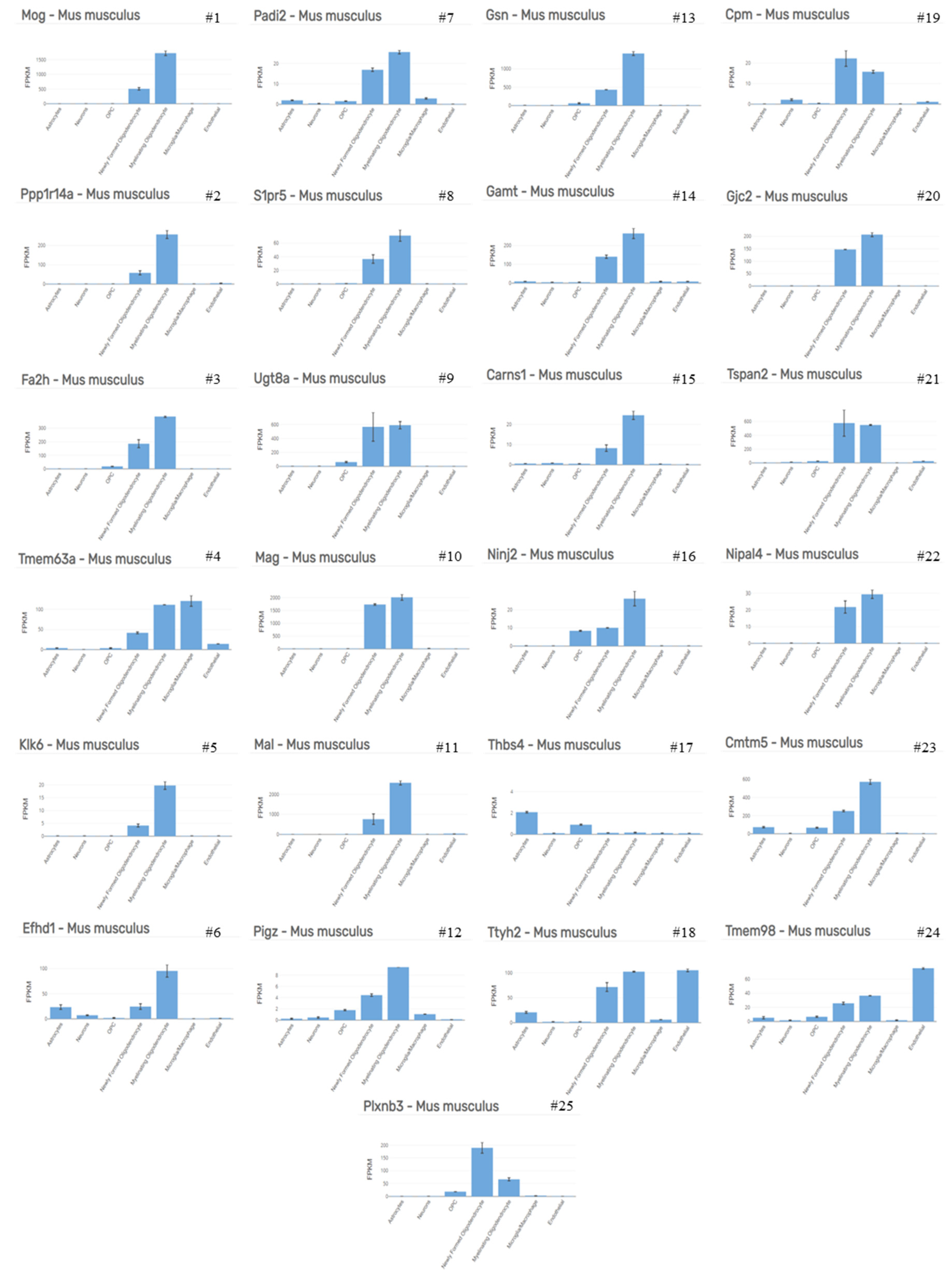
| Top 50 Up-Regulated Genes | Highest Expressed Cell Type | Top 50 Down-Regulated Genes | Highest Expressed Cell Type |
|---|---|---|---|
| Atf3 | Microglia/Macrophage | Mog | Myelinating Oligodendrocyte |
| Tgm1 | Microglia/Macrophage | Ppp1r14a | Myelinating Oligodendrocyte |
| Cxcl10 | Microglia/Macrophage | Fa2h | Myelinating Oligodendrocyte |
| Ccl3 | Microglia/Macrophage | Tmem63a | Microglia/Macrophage |
| Osmr | Endothelial | Klk6 | Myelinating Oligodendrocyte |
| Adamts1 | Endothelial | Efhd1 | Myelinating Oligodendrocyte |
| Hmox1 | Microglia/Macrophage | Padi2 | Myelinating Oligodendrocyte |
| Plscr2 | Astrocytes | S1pr5 | Myelinating Oligodendrocyte |
| Tnc | Astrocytes | Ugt8a | Myelinating oligodendrocyte |
| Cdkn1a | Endothelial | Mag | Myelinating oligodendrocyte |
| Serpina3n | OPC | Mal | Myelinating oligodendrocyte |
| Cd44 | Astrocytes | Pigz | Myelinating oligodendrocyte |
| Ddit3 | Microglia/Macrophage | Gsn | Myelinating oligodendrocyte |
| Serpinb1a | Myelinating oligodendrocyte | Gamt | Myelinating oligodendrocyte |
| Cyr61 | Astrocytes | Carns1 | Myelinating oligodendrocyte |
| Myc | Microglia/Macrophage | Ninj2 | Myelinating oligodendrocyte |
| Tnfrsf12a | Microglia/Macrophage | Thbs4 | Astrocytes |
| Fam46a | OPC | Ttyh2 | Endothelial |
| Slc14a1 | Astrocytes | Cpm | Newly formed oligodendrocyte |
| Fosl1 | Endothelial | Gjc2 | Myelinating oligodendrocyte |
| Dusp10 | Astrocytes | Tspan2 | Newly formed oligodendrocyte |
| Gadd45b | Microglia/Macrophage | Nipal4 | Myelinating oligodendrocyte |
| Clcf1 | Microglia/Macrophage | Cmtm5 | Myelinating oligodendrocyte |
| Gpr84 | Microglia/Macrophage | Tmem98 | Endothelial |
| Ccl2 | Microglia/Macrophage | Plxnb3 | Newly Formed Oligodendrocyte |
| A2m | Astrocytes | Pstpip2 | OPC |
| C3ar1 | Microglia/Macrophage | Slc15a2 | Astrocytes |
| Nupr1 | Microglia/Macrophage | Apln | Endothelial |
| Fos | Astrocytes | Ptgds | Newly formed oligodendrocyte |
| 1200009O22Rik | Unknown | Adssl1 | Myelinating oligodendrocyte |
| Ifrd1 | Microglia/Macrophage | Gstm7 | Endothelial |
| Gadd45g | Astrocytes | Apod | Myelinating oligodendrocyte |
| Arap2 | Astrocytes | Lrrn1 | Opc |
| Tgif1 | Microglia/Macrophage | Pllp | Newly formed oligodendrocyte |
| Ifit1 | Endothelial | Cntn2 | Myelinating oligodendrocyte |
| Lilrb4 | Microglia/Macrophage | Fah | Myelinating oligodendrocyte |
| Hbegf | Microglia/Macrophage | Serpind1 | Newly formed oligodendrocyte |
| Lcn2 | Microglia/Macrophage | Agt | Astrocytes |
| Ifit3 | Astrocytes | Anln | Myelinating oligodendrocyte |
| Myd116 | Microglia/Macrophage | Cryab | Myelinating oligodendrocyte |
| Stk40 | Microglia/Macrophage | Mboat1 | Myelinating oligodendrocyte |
| Trib3 | Endothelial | Kndc1 | Newly formed oligodendrocyte |
| Gbp2 | Endothelial | Lrp4 | Astrocytes |
| Myd88 | Microglia/Macrophage | Slc13a3 | Astrocytes |
| Tagln2 | Endothelial | Nmral1 | Myelinating oligodendrocyte |
| 1810010H24Rik | OPC | Fzd2 | Astrocytes |
| Slc1a5 | Endothelial | Paqr6 | Astrocytes |
| Phlda1 | OPC | Gja1 | Astrocytes |
| Egr2 | Microglia/Macrophage | Scd1 | Myelinating oligodendrocyte |
| Slc7a11 | Astrocytes | Fam57a | Myelinating oligodendrocyte |
| Histopathology of Progressive MS | Reference | Cuprizone Model | Reference |
|---|---|---|---|
| Gray matter demyelination | [5,76] | Demyelination in cortical and subcortical structures | [77,78] |
| Diffuse white matter damage | [76,79] | Demyelination of white matter tracts, especially in the corpus callosum | [78,80] |
| Axonal damage | [81,82,83] | Axonal damage in demyelinated areas | [56,84] |
| Minor immune cell infiltration | [85,86] | Little or no infiltration of lymphocytes | [87,88] |
| Minor BBB integrity loss | [89,90,91] | Minor BBB integrity loss | [22,23] |
| Profound oxidative injury | [92,93,94] | Accumulation of oxidative damage | [95,96] |
| Progressive worsening of function | [97,98] | Impaired motor coordination, spatial memory and social behavior | [22,50,65,66,67,68,69,70,72,73] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, J.; Mann, T.; Joost, S.; Behrangi, N.; Frank, M.; Kipp, M. The Cuprizone Model: Dos and Do Nots. Cells 2020, 9, 843. https://doi.org/10.3390/cells9040843
Zhan J, Mann T, Joost S, Behrangi N, Frank M, Kipp M. The Cuprizone Model: Dos and Do Nots. Cells. 2020; 9(4):843. https://doi.org/10.3390/cells9040843
Chicago/Turabian StyleZhan, Jiangshan, Teresa Mann, Sarah Joost, Newshan Behrangi, Marcus Frank, and Markus Kipp. 2020. "The Cuprizone Model: Dos and Do Nots" Cells 9, no. 4: 843. https://doi.org/10.3390/cells9040843
APA StyleZhan, J., Mann, T., Joost, S., Behrangi, N., Frank, M., & Kipp, M. (2020). The Cuprizone Model: Dos and Do Nots. Cells, 9(4), 843. https://doi.org/10.3390/cells9040843