Targeting Autophagy Augments Berberine-Mediated Cell Death in Human Hepatoma Cells Harboring Hepatitis C Virus RNA

 , ,
, ,
Abstract
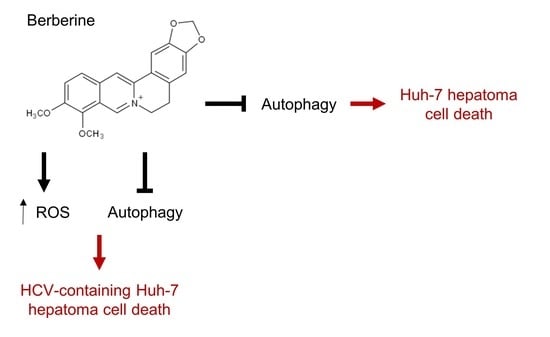
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
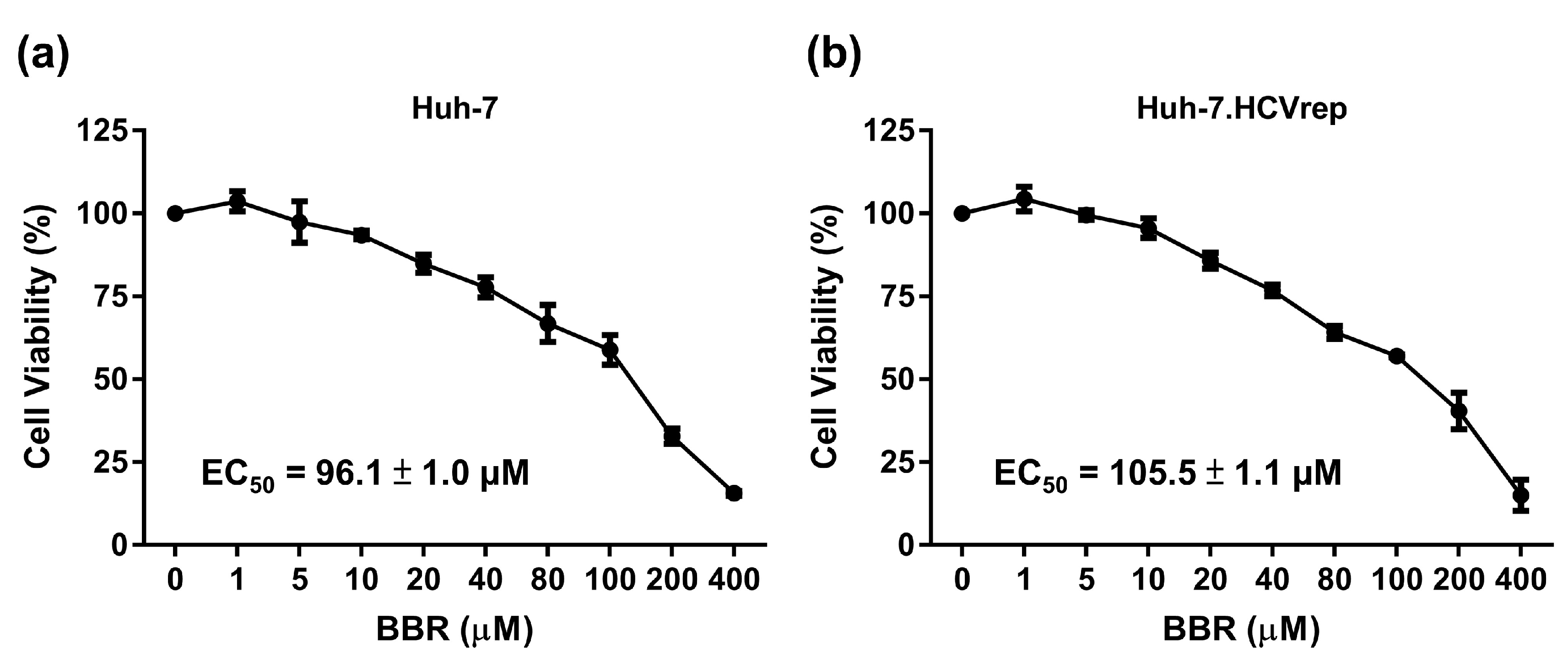
2.1. BBR Inhibits Hepatoma Cell Growth Irrespective of HCV RNA Expression
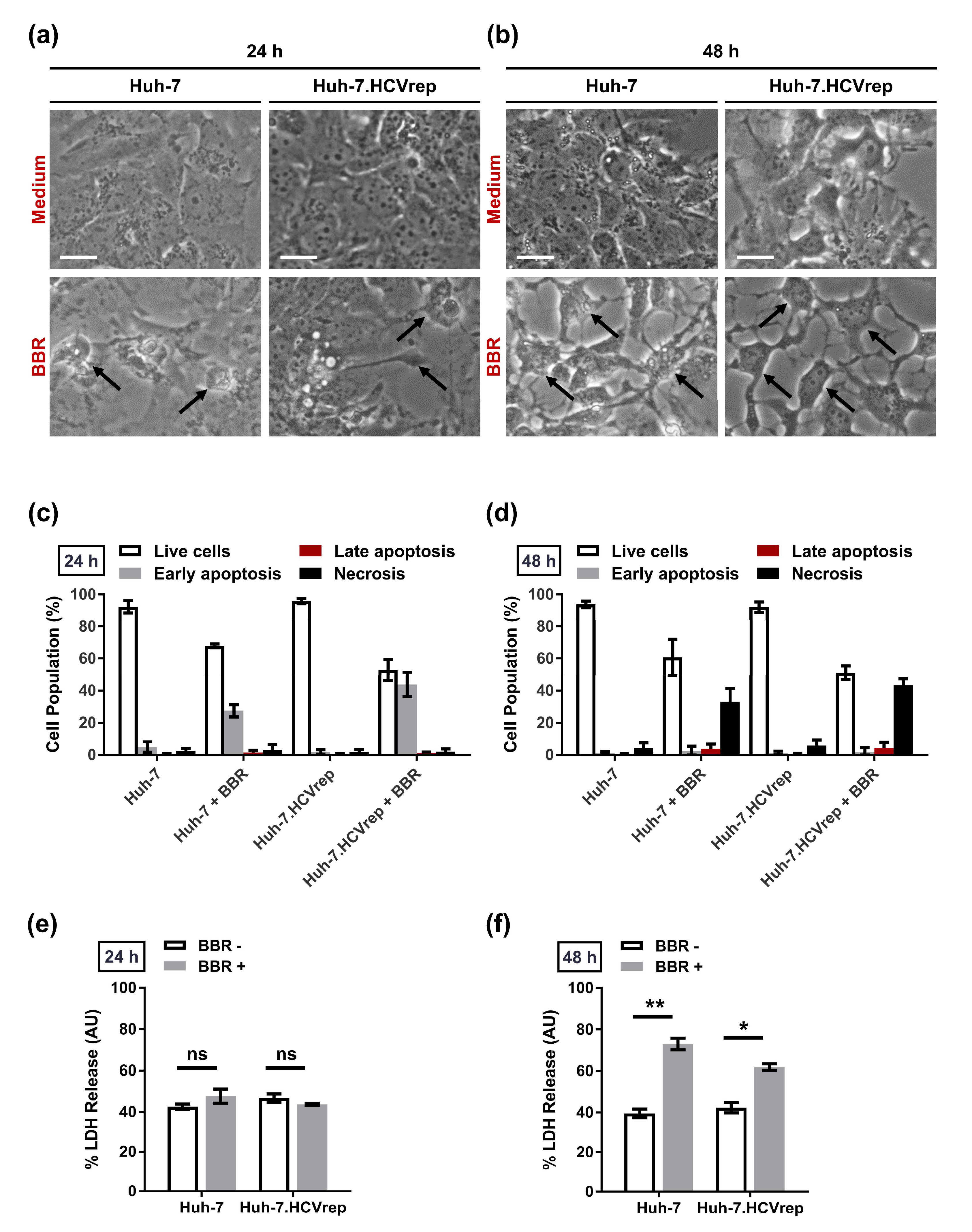
2.2. BBR Induces Time-Dependent Biphasic Hepatoma Cell Death
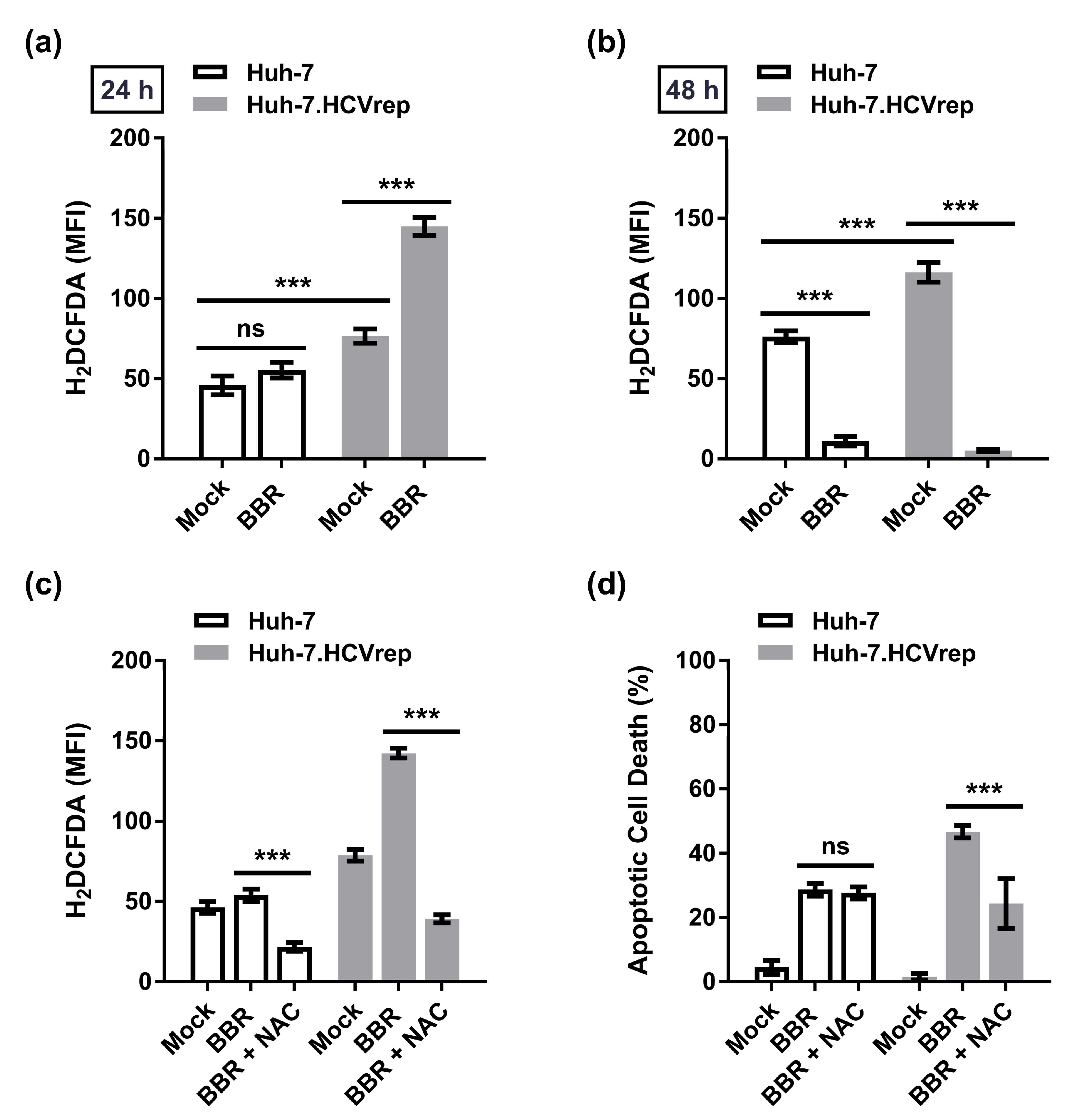
2.3. Inhibition of ROS Attenuates the BBR-Induced HCV Replicon Cell Death, But Not the Parental HCV RNA-Negative Huh-7 Cells
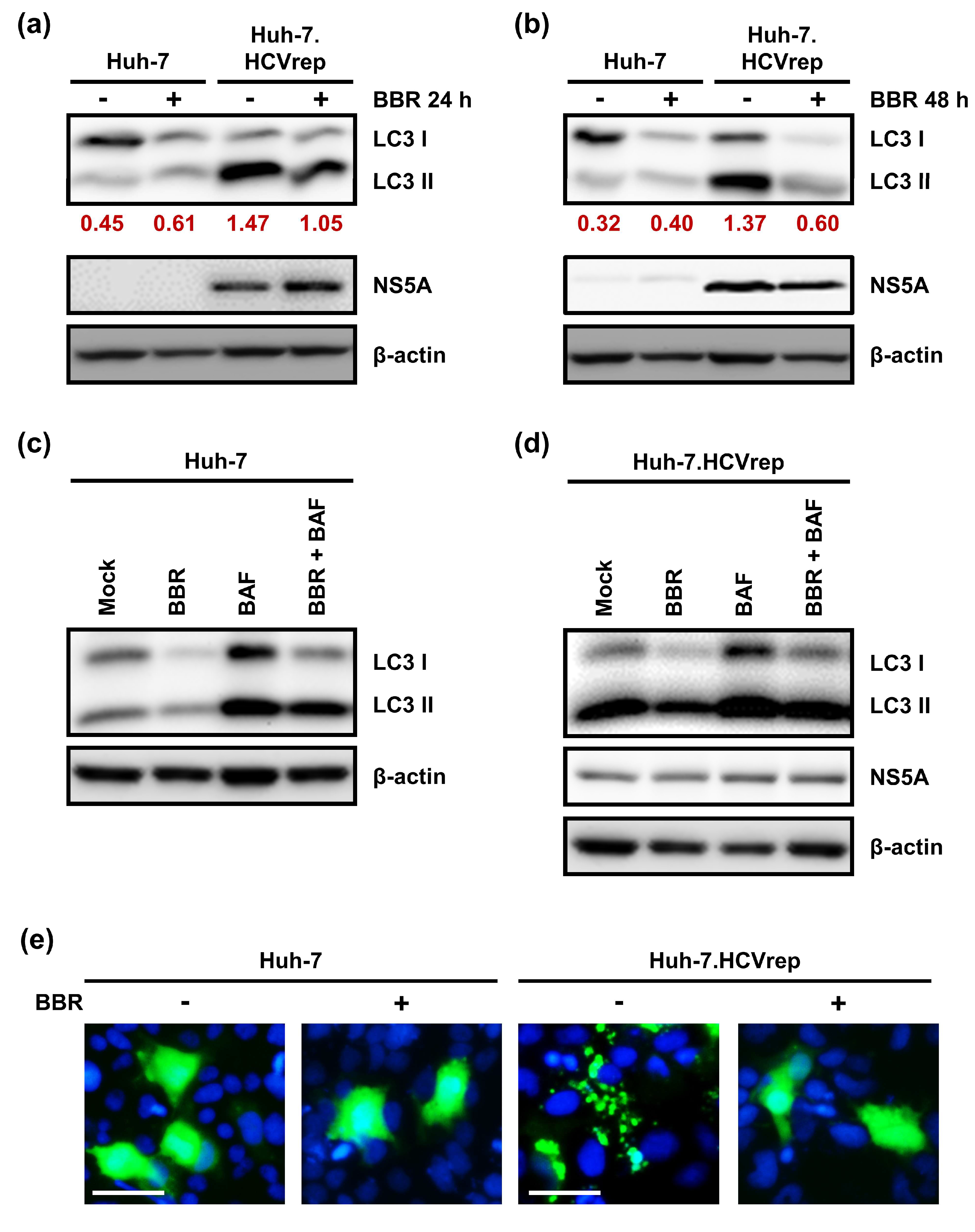
2.4. BBR Modulates Autophagy in HCC Cells
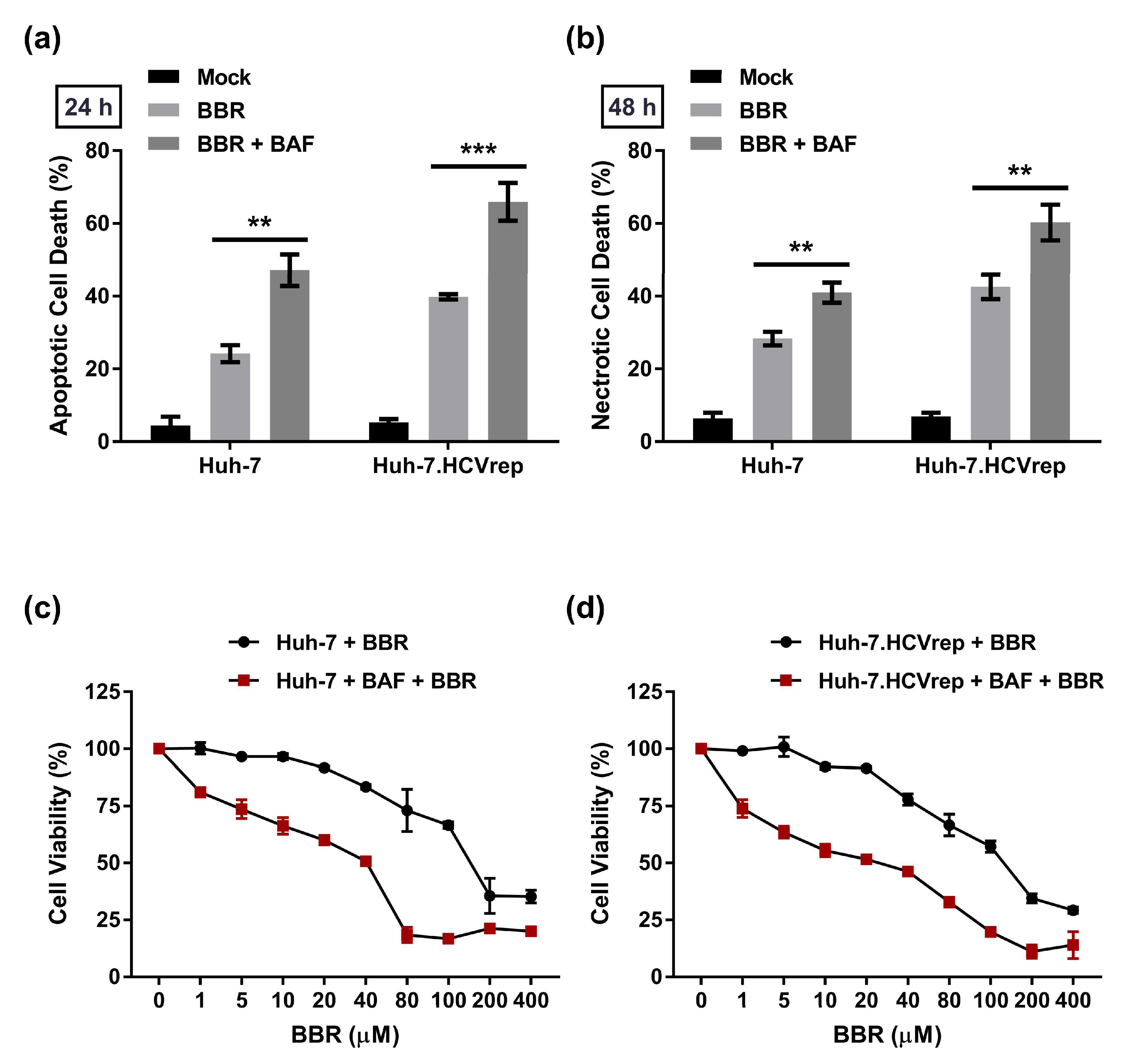
2.5. BAF Pretreatment Augments BBR-Induced Cell Death in Hepatoma Cells Irrespective of Intracellular HCV RNA Replication
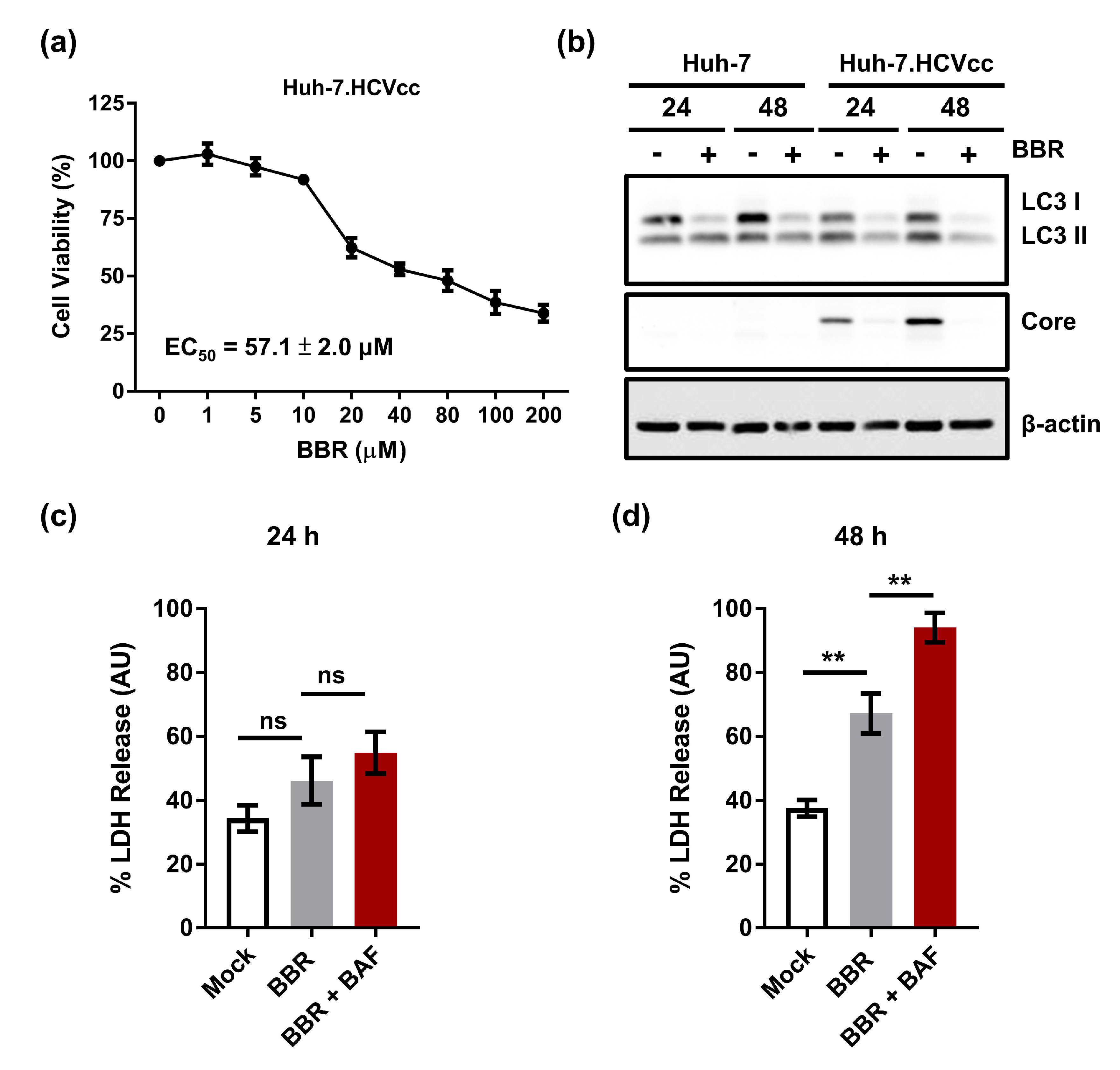
2.6. Inhibition of Autophagy Enhances BBR-Induced Death of Cells Persistently Infected With HCV Particles
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cytotoxicity Assay
4.4. Analysis of Cell Death
4.5. Reactive Oxygen Species Production and Scavenging Analysis
4.6. Bafilomycin A1 Treatment
4.7. Lactose Dehydrogenase Activity Assay for the Detection of Necrosis
4.8. Western Blotting
4.9. EGFP-LC3 Fluorescence Microscopy
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Elbedewy, T.A.; El-Serafy, M.; El-Toukhy, N.; Ahmed, W.; Ali El Din, Z. Hepatitis C virus: A global view. World J. Hepatol. 2015, 7, 2676–2680. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M.; American Association for the Study of Liver, D. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ducreux, M.; Lencioni, R.; Di Bisceglie, A.; Galle, P.; Dufour, J. European Association For The Study Of The Liver European Organisation For Research And Treatment Of Cancer: EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar]
- Deng, G.L.; Zeng, S.; Shen, H. Chemotherapy and target therapy for hepatocellular carcinoma: New advances and challenges. World J. Hepatol. 2015, 7, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Ohtsuki, T.; Obata, H.; Tomimatsu, M.; Okazaki, N.; Hasegawa, H.; Nakajima, Y.; Ohnishi, K. Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer 1985, 56, 918–928. [Google Scholar] [CrossRef]
- Butt, A.S.; Sharif, F.; Abid, S. Impact of direct acting antivirals on occurrence and recurrence of hepatocellular carcinoma: Biologically plausible or an epiphenomenon? World J. Hepatol. 2018, 10, 267–276. [Google Scholar] [CrossRef]
- Zoulim, F.; Liang, T.J.; Gerbes, A.L.; Aghemo, A.; Deuffic-Burban, S.; Dusheiko, G.; Fried, M.W.; Pol, S.; Rockstroh, J.K.; Terrault, N.A. Hepatitis C virus treatment in the real world: Optimising treatment and access to therapies. Gut 2015, 64, 1824–1833. [Google Scholar] [CrossRef] [Green Version]
- Villani, R.; Vendemiale, G.; Serviddio, G. Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy. Int. J. Mol. Sci. 2019, 20, 49. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Wang, N.; Zhao, L.; Lu, F. Berberine in the treatment of type 2 diabetes mellitus: A systemic review and meta-analysis. Evid. Based Complement. Alternat. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Feng, Y.; Tsao, S.; Wang, N.; Curtain, R.; Wang, Y. Berberine and Coptidis rhizoma as novel antineoplastic agents: A review of traditional use and biomedical investigations. J. Ethnopharm. 2009, 126, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Feng, Y.; Zhu, M.; Tsang, C.M.; Man, K.; Tong, Y.; Tsao, S.W. Berberine induces autophagic cell death and mitochondrial apoptosis in liver cancer cells: The cellular mechanism. J. Cell Biochem. 2010, 111, 1426–1436. [Google Scholar] [CrossRef] [Green Version]
- Yip, N.K.; Ho, W.S. Berberine induces apoptosis via the mitochondrial pathway in liver cancer cells. Oncol. Rep. 2013, 30, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Zhang, Z.-Q.; Wang, B.; Jiang, H.-X.; Cheng, L.; Shen, L.-M. Berberine-induced apoptotic and autophagic death of HepG2 cells requires AMPK activation. Cancer Cell Int. 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.L.; Jackson, W.T. How positive-strand RNA viruses benefit from autophagosome maturation. J. Virol. 2013, 87, 9966–9972. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ou, J.-H.J. Hepatitis C virus and autophagy. Biol. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xun, K.; Wang, Y.; Chen, X. A systematic review of the anticancer properties of berberine, a natural product from Chinese herbs. Anticancer Drugs 2009, 20, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [PubMed] [Green Version]
- Burd, J.F.; Usategui-Gomez, M. A colorimetric assay for serum lactate dehydrogenase. Clin. Chim. Acta 1973, 46, 223–227. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2’, 7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses 2016, 8, 150. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Wileman, T.J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- White, E. Autophagic cell death unraveled: Pharmacological inhibition of apoptosis and autophagy enables necrosis. Autophagy 2008, 4, 399–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Ye, L.; Bai, Y.; Sun, A.; Cox, B.; Liu, D.; Li, Y.; Liotta, D.; Snyder, J.P.; Fu, H. Autophagy and apoptosis in hepatocellular carcinoma induced by EF25-(GSH) 2: A novel curcumin analog. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Zhuang, J.; Ji, C.; Cai, Z.; Liao, W.; Huang, Z. Curcumin inhibits hepatocellular carcinoma growth by targeting VEGF expression. Oncol. Lett. 2018, 15, 4821–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishayee, A.; Politis, T.; Darvesh, A.S. Resveratrol in the chemoprevention and treatment of hepatocellular carcinoma. Cancer Treat. Rev. 2010, 36, 43–53. [Google Scholar] [CrossRef]
- Lin, C.; Chang, T.; Hsieh, W.; Hung, M.; Lin, I.; Lai, S.; Tzeng, Y. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Discov. 2016, 2, 16065. [Google Scholar] [CrossRef] [Green Version]
- Varghese, L.; Agarwal, C.; Tyagi, A.; Singh, R.P.; Agarwal, R. Silibinin efficacy against human hepatocellular carcinoma. Clin. Cancer Res. 2005, 11, 8441–8448. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly (ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Leist, M.; Single, B.; Castoldi, A.F.; Kühnle, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef]
- Xu, M.; Xiao, Y.; Yin, J.; Hou, W.; Yu, X.; Shen, L.; Liu, F.; Wei, L.; Jia, W. Berberine promotes glucose consumption independently of AMP-activated protein kinase activation. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, O.A.; Ivanova, O.N.; Bartosch, B.; Valuev-Elliston, V.T.; Mukhtarov, F.; Kochetkov, S.N.; Ivanov, A.V. Hepatitis C Virus NS5A Protein Triggers Oxidative Stress by Inducing NADPH Oxidases 1 and 4 and Cytochrome P450 2E1. Oxid. Med. Cell Longev. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-κB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeran, S.M.; Katiyar, S.; Katiyar, S.K. Berberine-induced apoptosis in human prostate cancer cells is initiated by reactive oxygen species generation. Toxicol. Appl. Pharmacol. 2008, 229, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Mandal, M.; Kögel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766. [Google Scholar] [CrossRef]
- Hung, T.-C.; Jassey, A.; Liu, C.-H.; Lin, C.-J.; Lin, C.-C.; Wong, S.H.; Wang, J.Y.; Yen, M.-H.; Lin, L.-T. Berberine inhibits hepatitis C virus entry by targeting the viral E2 glycoprotein. Phytomedicine 2019, 53, 62–69. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, J.; Zhang, X.; Yang, J.; Zhang, D.; Huang, J.; Lv, P.; Shen, W.; Yang, Y. Berberine attenuates autophagy in adipocytes by targeting BECN1. Autophagy 2014, 10, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Jassey, A.; Liu, C.H.; Changou, C.A.; Richardson, C.D.; Hsu, H.Y.; Lin, L.T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.H.; Lin, C.C.; Hsu, W.C.; Chung, C.Y.; Lin, C.C.; Jassey, A.; Chang, S.P.; Tai, C.J.; Tai, C.J.; Shields, J.; et al. Highly bioavailable silibinin nanoparticles inhibit HCV infection. Gut 2017, 66, 1853–1861. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tai, C.-J.; Jassey, A.; Liu, C.-H.; Tai, C.-J.; Richardson, C.D.; Wong, S.H.; Lin, L.-T. Targeting Autophagy Augments Berberine-Mediated Cell Death in Human Hepatoma Cells Harboring Hepatitis C Virus RNA. Cells 2020, 9, 908. https://doi.org/10.3390/cells9040908
Tai C-J, Jassey A, Liu C-H, Tai C-J, Richardson CD, Wong SH, Lin L-T. Targeting Autophagy Augments Berberine-Mediated Cell Death in Human Hepatoma Cells Harboring Hepatitis C Virus RNA. Cells. 2020; 9(4):908. https://doi.org/10.3390/cells9040908
Chicago/Turabian StyleTai, Chen-Jei, Alagie Jassey, Ching-Hsuan Liu, Cheng-Jeng Tai, Christopher D. Richardson, Shu Hui Wong, and Liang-Tzung Lin. 2020. "Targeting Autophagy Augments Berberine-Mediated Cell Death in Human Hepatoma Cells Harboring Hepatitis C Virus RNA" Cells 9, no. 4: 908. https://doi.org/10.3390/cells9040908
APA StyleTai, C. -J., Jassey, A., Liu, C. -H., Tai, C. -J., Richardson, C. D., Wong, S. H., & Lin, L. -T. (2020). Targeting Autophagy Augments Berberine-Mediated Cell Death in Human Hepatoma Cells Harboring Hepatitis C Virus RNA. Cells, 9(4), 908. https://doi.org/10.3390/cells9040908