CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review

Abstract
:1. Introduction
1.1. CRISPR Epigenome Editors
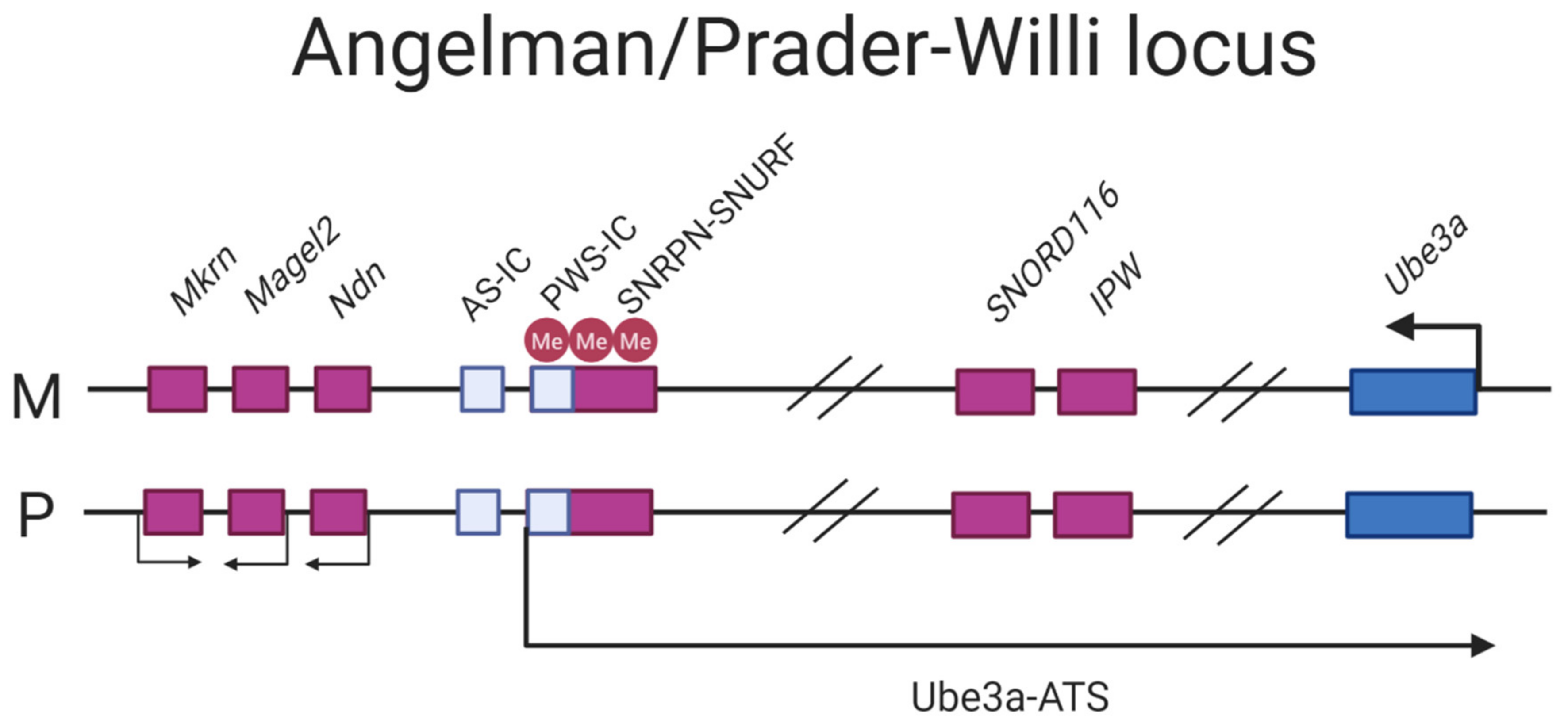
1.2. Genomic Imprinting, Rare Imprinting Diseases, and Epigenome Engineering
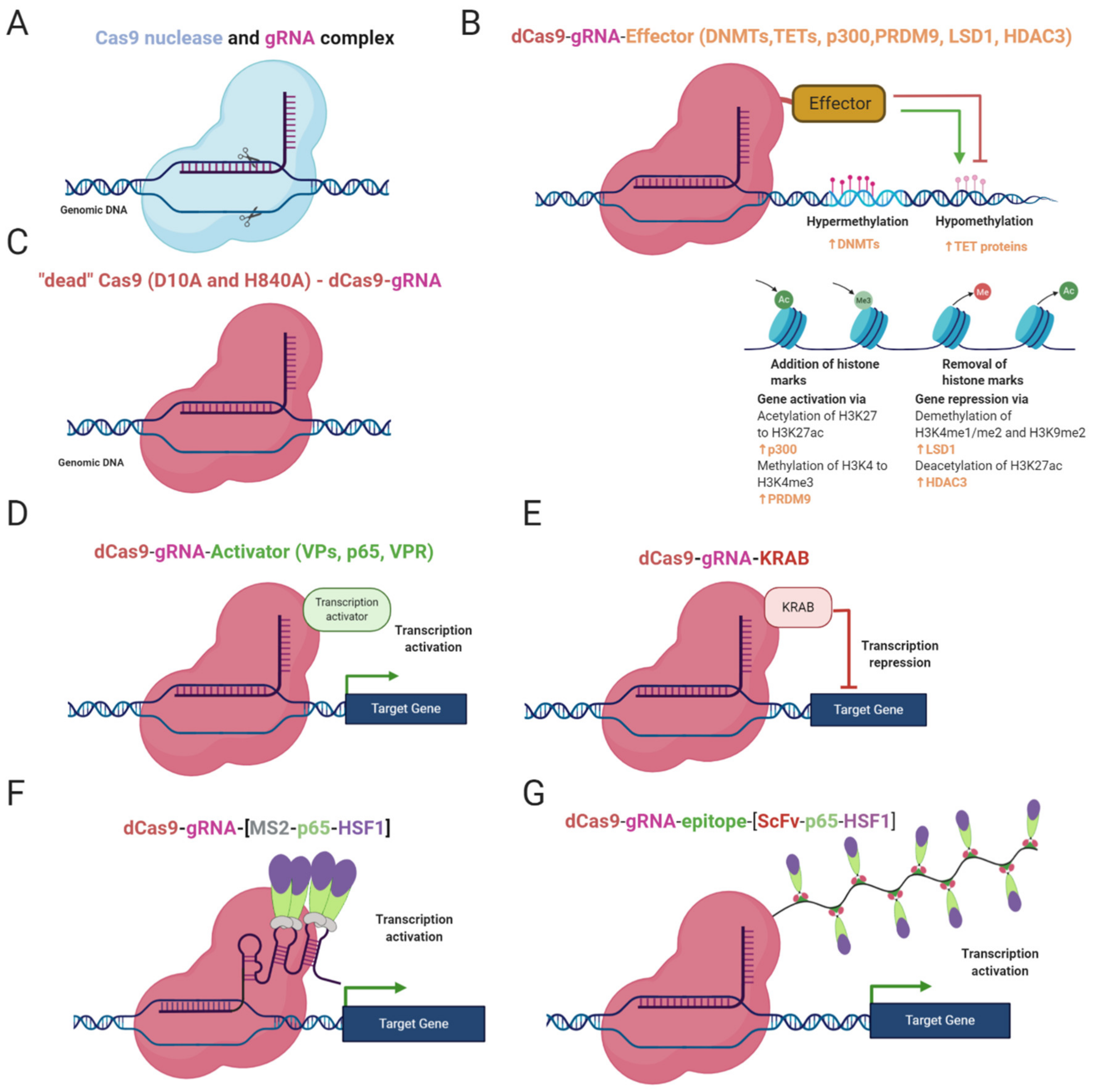
2. CRISPR-Based Epigenome Editors (CRISPR Epi-Editors)
2.1. DNA De/Methylation Mediated by CRISPR Epigenome Editors
2.2. Histone Modifications by CRISPR Epi-Editors
2.3. Gene Regulation by CRISPRa and CRISPRi Systems
2.4. Delivery
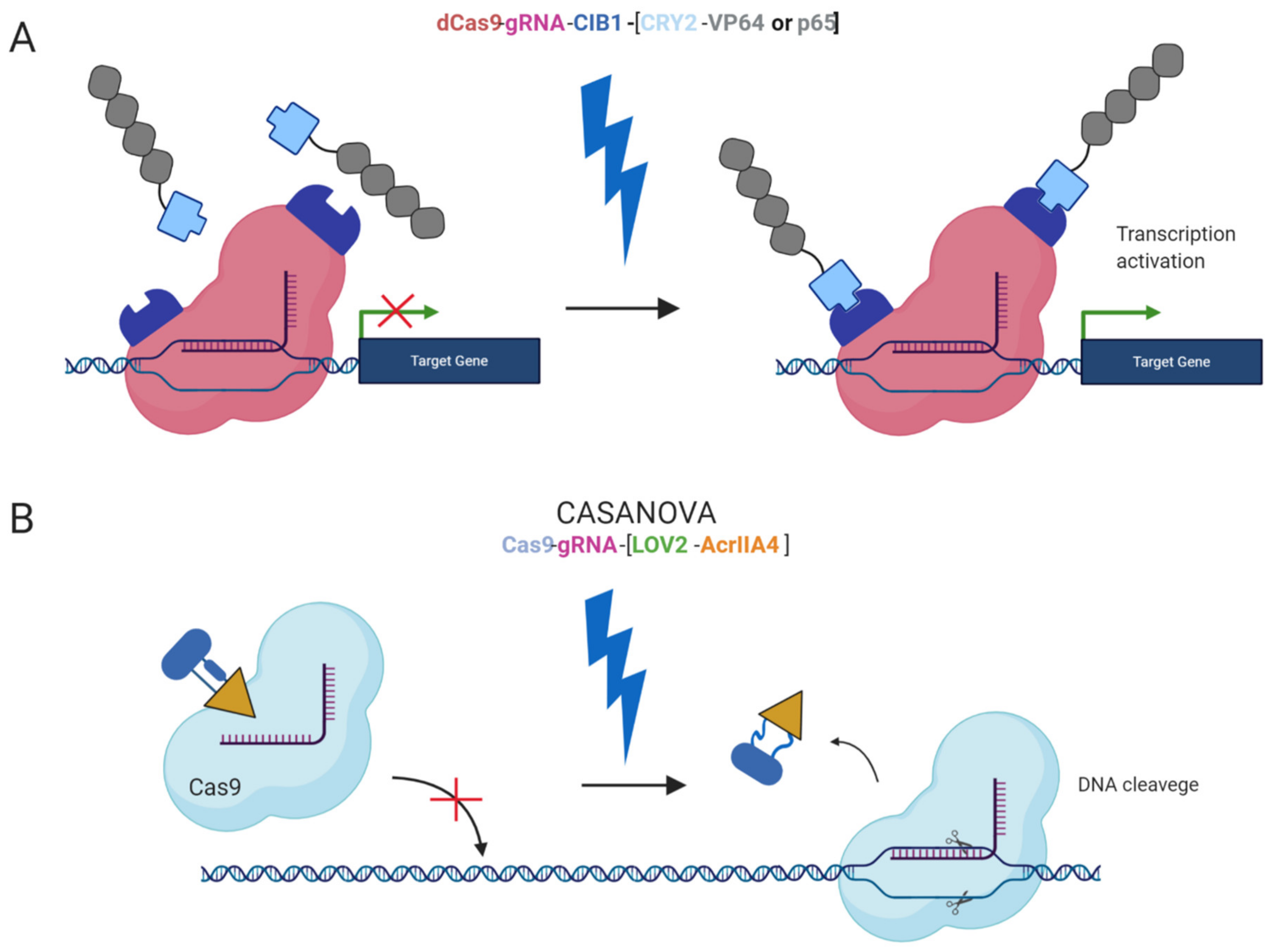
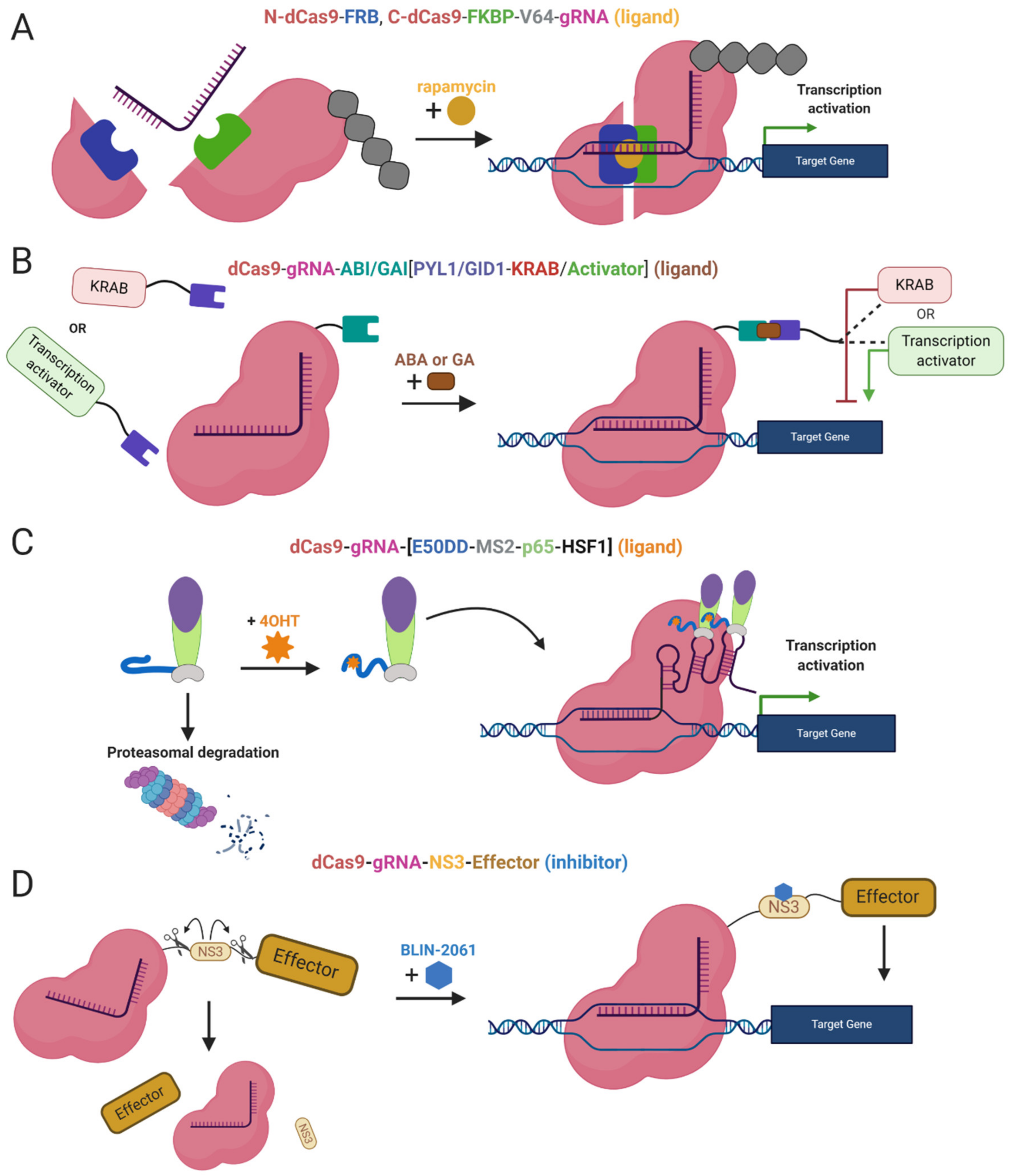
2.5. Inducible Systems
3. Rare Imprinting Diseases and Therapy
3.1. Angelman Syndrome
3.2. Prader–Willi Syndrome
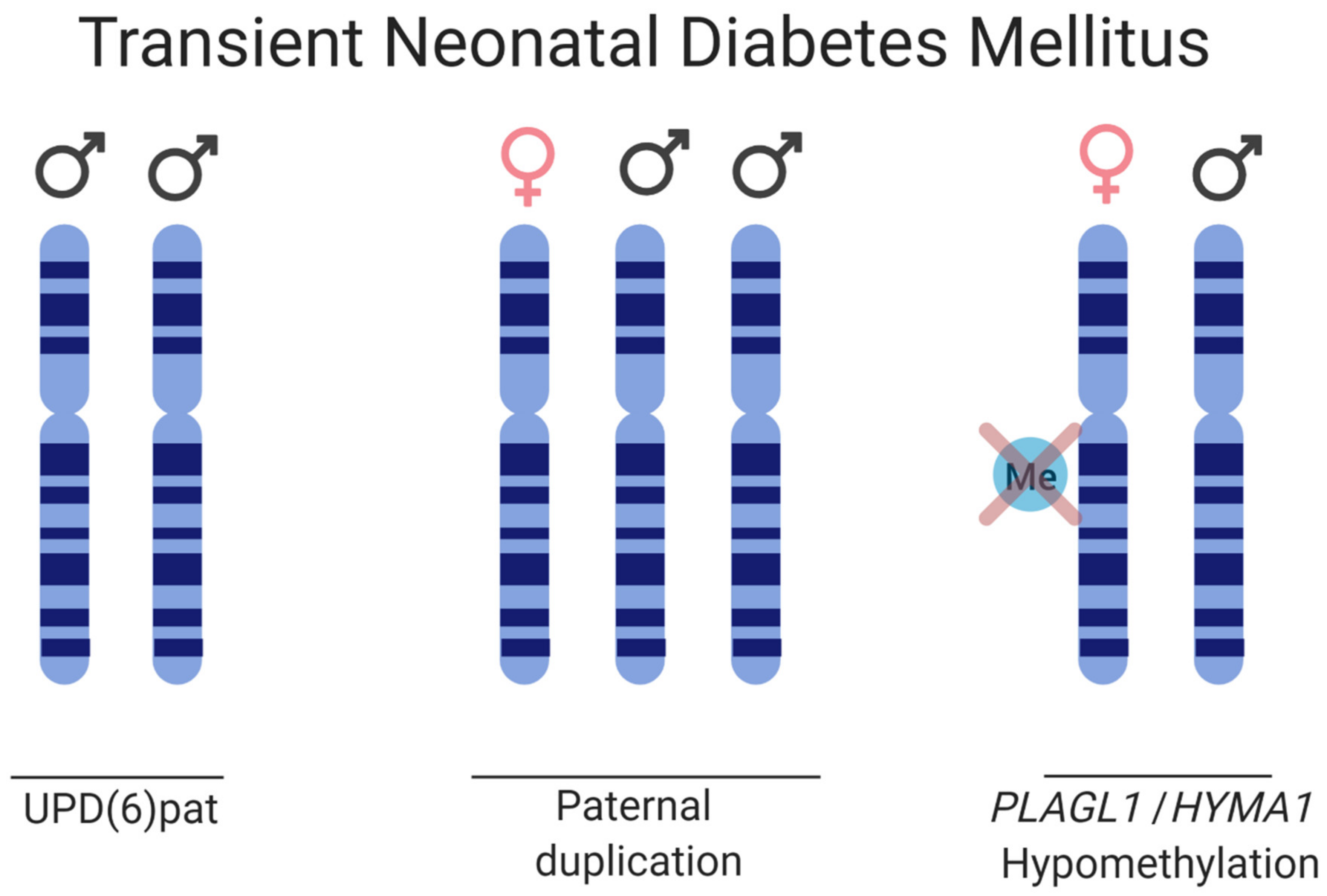
3.3. Transient Neonatal Diabetes Mellitus Type 1
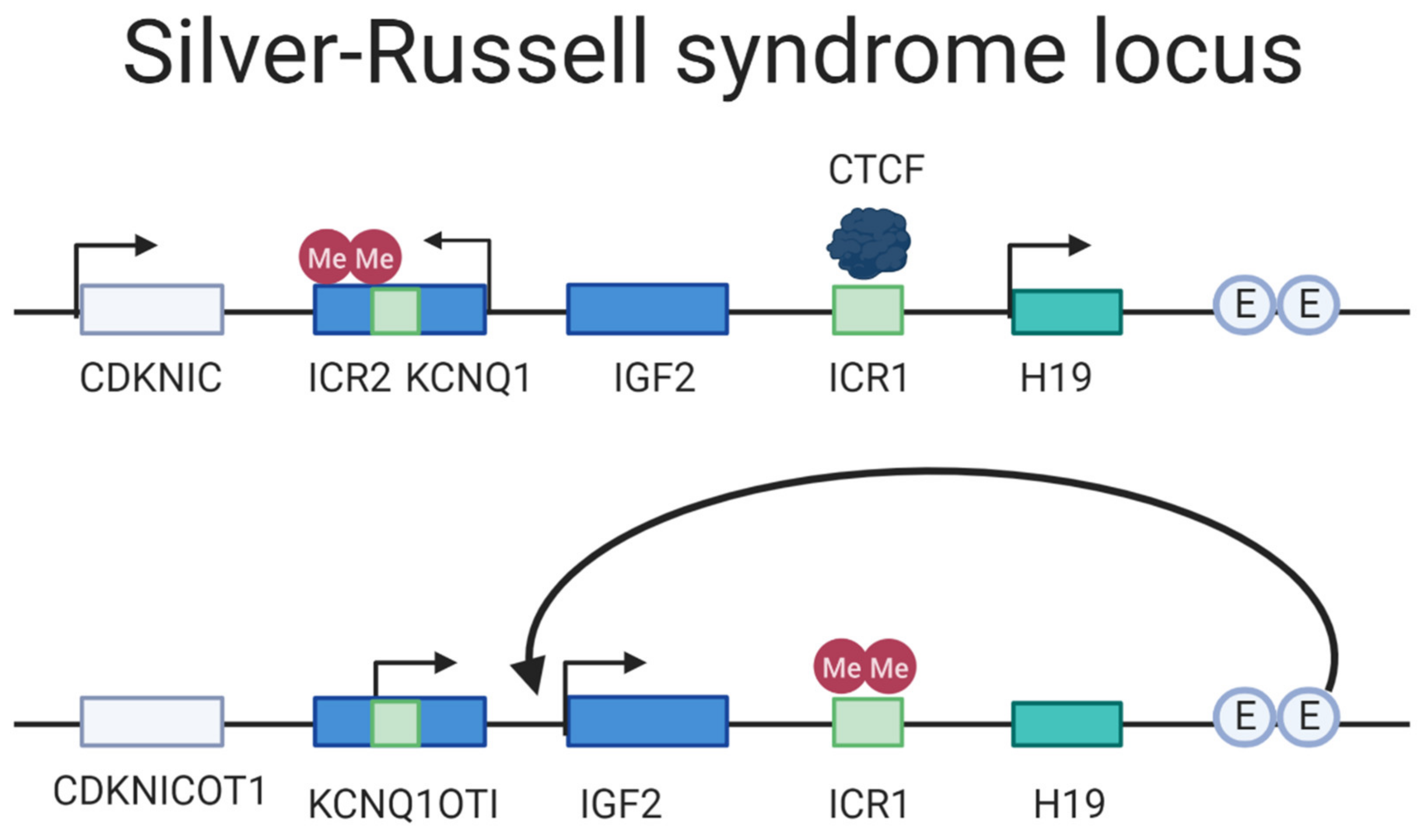
3.4. Silver–Russell Syndrome
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Hara, S.; Goto, Y.; Ogawa, Y.; Okayasu, H.; Kubota, S.; Tamano, M.; Terao, M.; Takada, S. Creation of mutant mice with megabase-sized deletions containing custom-designed breakpoints by means of the CRISPR/Cas9 system. Sci. Rep. 2017, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Kitamoto, K.; Taketani, Y.; Fujii, W.; Inamochi, A.; Toyono, T.; Miyai, T.; Yamagami, S.; Kuroda, M.; Usui, T.; Ouchi, Y. Generation of mouse model of TGFBI-R124C corneal dystrophy using CRISPR/Cas9-mediated homology-directed repair. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Tasca, F.; Wang, Q.; Liu, J.; Janssen, J.M.; Brescia, M.D.; Bellin, M.; Szuhai, K.; Kenrick, J.; Frock, R.L.; et al. Expanding the editable genome and CRISPR-Cas9 versatility using DNA cutting-free gene targeting based on in trans paired nicking. Nucleic Acids Res. 2020, 48, 974–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigouroux, A.; Oldewurtel, E.; Cui, L.; Bikard, D.; Teeffelen, S. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol. 2018, 14. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef]
- Inbar-Feigenberg, M.; Choufani, S.; Butcher, D.T.; Roifman, M.; Weksberg, R. Basic concepts of epigenetics. Fertil. Steril. 2013, 99, 607–615. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Bourc’his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadakierska-Chudy, A.; Kostrzewa, R.M.; Filip, M. A Comprehensive View of the Epigenetic Landscape Part I: DNA Methylation, Passive and Active DNA Demethylation Pathways and Histone Variants. Neurotox. Res. 2015, 27, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Hill, P.W.S.; Amouroux, R.; Hajkova, P. DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: An emerging complex story. Genomics 2014, 104, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Coz, M.L.; Devarajan, K.; Wessels, A.; Soprano, D.; Abramowitz, K.; et al. Demethylation by Linked Deamination-Base Excision Repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Dizdaroglu, M.; Bergtold, D.S. Characterization of free radical-induced base damage in DNA at biologically relevant levels. Anal. Biochem. 1986, 156, 182–188. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.M.; Andrews, R.M.; Flicek, P.; Dillon, S.C.; Karaöz, U.; Clelland, G.K.; Wilcox, S.; Beare, D.M.; Fowler, J.C.; Couttet, P.; et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007, 17, 691–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, K.; Yin, L.; Yu, Y.; Qi, J.; Shen, W.H.; Zhu, J.; Zhang, Y.; Dong, A. H3K4me2 functions as a repressive epigenetic mark in plants. Epigenet. Chromatin 2019, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Wang, Z.; Schones, D.E.; Zhao, K.; DeSalle, R.; Zhang, M.Q. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genom. 2009, 10, 143. [Google Scholar] [CrossRef] [Green Version]
- Lu, D. Epigenetic modification enzymes: Catalytic mechanisms and inhibitors. Acta Pharm. Sin. B 2013, 3, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Enríquez, P. CRISPR-mediated epigenome editing. Yale J. Biol. Med. 2016, 89, 471–486. [Google Scholar]
- Surani, M.A.H.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.M.; Thomas, S.; Beechey, C.V.; Hancock, J.; Cattanach, B.M.; Peters, J. MRC Harwell, Oxfordshire. World Wide Web Site - Mouse Imprinting Data and References. 2013. Available online: http://www.har.mrc.ac.uk/research/genomic_imprinting/ (accessed on 3 March 2020).
- Chotalia, M.; Smallwood, S.A.; Ruf, N.; Dawson, C.; Lucifero, D.; Frontera, M.; James, K.; Dean, W.; Kelsey, G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009, 23, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, D.P. Genomic Imprinting: A Mammalian Epigenetic Discovery Model. Annu. Rev. Genet. 2011, 45, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Person, R.E.; Beaudet, A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012, 21, 3001–3012. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneda, M.; Hata, K.; Kumaki, K.; Hisano, M.; Kohara, Y.; Okano, M.; Li, E.; Nozaki, M.; Sasaki, H. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum. Mol. Genet. 2007, 16, 2272–2280. [Google Scholar] [CrossRef]
- Russo, S.; Calzari, L.; Mussa, A.; Mainini, E.; Cassina, M.; Di Candia, S.; Clementi, M.; Guzzetti, S.; Tabano, S.; Miozzo, M.; et al. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver–Russell and Beckwith–Wiedemann syndromes. Clin. Epigenetics 2016, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, J.F.; Úbeda, F. Chapter 13—Diseases Associated with Genomic Imprinting. In Progress in Molecular Biology and Translational Science; Cheng, X., Blumenthal, R.M., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 101, pp. 401–445. ISBN 1877-1173. [Google Scholar]
- Eggermann, T.; Netchine, I.; Temple, I.K.; Tümer, Z.; Monk, D.; Mackay, D.; Grønskov, K.; Riccio, A.; Linglart, A.; Maher, E.R. Congenital imprinting disorders: EUCID.net—A network to decipher their aetiology and to improve the diagnostic and clinical care. Clin. Epigenetics 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenetics 2014, 1, 9–19. [Google Scholar] [CrossRef]
- Ptak, C.; Petronis, A. Epigenetics and Complex Disease: From Etiology to New Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 257–276. [Google Scholar] [CrossRef]
- El Bahhaj, F.; Dekker, F.J.; Martinet, N.; Bertrand, P. Delivery of epidrugs. Drug Discov. Today 2014, 19, 1337–1352. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Núñez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; P R Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442. [Google Scholar] [CrossRef] [Green Version]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, H.; Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Grieger, J.C.; Samulski, R.J. Packaging Capacity of Adeno-Associated Virus Serotypes: Impact of Larger Genomes on Infectivity and Postentry Steps. J. Virol. 2005, 79, 9933–9944. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.F.; Lázaro-Ibáñez, E.; Forsgard, M.A.M.; Shatnyeva, O.; Osteikoetxea, X.; Karlsson, F.; Heath, N.; Ingelsten, M.; Rose, J.; Harris, J.; et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 2019, 11, 6990–7001. [Google Scholar] [CrossRef]
- Matharu, N.; Rattanasopha, S.; Tamura, S.; Maliskova, L.; Wang, Y.; Bernard, A.; Hardin, A.; Eckalbar, W.L.; Vaisse, C.; Ahituv, N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363, eaau0629. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Fitzpatrick, Z.; Crommentuijn, M.H.W.; Mu, D.; Maguire, C.A. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials 2014, 35, 7598–7609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, S.M.G.; Kirkland, J.G.; Chory, E.J.; Husmann, D.; Calarco, J.P.; Crabtree, G.R. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- MacLeod, R.S.; Cawley, K.M.; Gubrij, I.; Nookaew, I.; Onal, M.; O’Brien, C.A. Effective CRISPR interference of an endogenous gene via a single transgene in mice. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [Green Version]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Cano-Rodriguez, D.; Gjaltema, R.A.F.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.J.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S.; Yuan, X.; Rots, M.G.; Strahl, B.D.; Blancafort, P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012, 7, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, D.L.; Le Lay, J.E.; Ruano, E.G.; Kaestner, K.H. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J. Clin. Invest. 2015, 125, 1998–2006. [Google Scholar] [CrossRef]
- Chen, H.; Kazemier, H.G.; De Groote, M.L.; Ruiters, M.H.J.; Xu, G.-L.; Rots, M.G. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014, 42, 1563–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Lo, A. Genetic and epigenetic control of gene expression by CRISPR-Cas systems. F1000Research 2017, 6, 747. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Senanayake, U.; Artibani, M.; Taylor, G.; Wells, D.; Ahmed, A.A.; Sauka-Spengler, T. Genome and epigenome engineering CRISPR toolkit for in vivo modulation of cis-regulatory interactions and gene expression in the chicken embryo. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Tani, T.; Kikyo, N. Structure and functions of powerful transactivators: VP16, MyoD and FoxA. Int. J. Dev. Biol. 2011, 54, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparison of Cas9 activators in multiple species. Nat. Methods 2016, 13, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S.; et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019, 176, 377–390.e19. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liu, Y.; Cao, H.; Zhang, Y.; Gu, Z.; Liu, X.; Yu, A.; Kaphle, P.; Dickerson, K.E.; Ni, M.; et al. Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Connoly, J.B. Lentiviruses in gene therapy clinical reseach. Gene Ther. 2002, 9, 1730–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Muñoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.; Toth, K. Adenovirus Vectors for Gene Therapy, Vaccination and Cancer Gene Therapy. Curr. Gene Ther. 2014, 13, 421–433. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017, 35, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieger, J.C.; Choi, V.W.; Samulski, R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006, 1, 1412–1428. [Google Scholar] [CrossRef]
- Hudry, E.; Andres-Mateos, E.; Lerner, E.P.; Volak, A.; Cohen, O.; Hyman, B.T.; Maguire, C.A.; Vandenberghe, L.H. Efficient Gene Transfer to the Central Nervous System by Single-Stranded Anc80L65. Mol. Ther. Methods Clin. Dev. 2018, 10, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.D.; Aschenbrenner, S.; Grosse, S.; Rapti, K.; Domenger, C.; Fakhiri, J.; Mastel, M.; Orner, K.B.; Eils, R.; Grimm, D.; et al. Cell-specific CRISPR-Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res. 2019, 47, 75. [Google Scholar] [CrossRef] [Green Version]
- Thakore, P.I.; Kwon, J.B.; Nelson, C.E.; Rouse, D.C.; Gemberling, M.P.; Oliver, M.L.; Gersbach, C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.W.; Mali, P.; Wu, E.Y.; Ng, A.H.M.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016. [Google Scholar] [CrossRef] [Green Version]
- Vogt, S.; Stadlmayr, G.; Grillari, J.; Rüker, F.; Wozniak-Knopp, G. Engineering of Surface Proteins in Extracellular Vesicles for Tissue-Specific Targeting. In Current Topics in Biochemical Engineering; IntechOpen: London UK, 2019. [Google Scholar]
- Göran Ronquist, K. Extracellular vesicles and energy metabolism. Clin. Chim. Acta 2019, 488, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Van Gestel, M.A.; Boender, A.J.; De Vrind, V.A.J.; Garner, K.M.; Luijendijk, M.C.M.; Adan, R.A.H. Recombinant adeno-associated virus: Efficient transduction of the rat VMH and clearance from blood. PLoS ONE 2014, 9, e97639. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.W.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [Green Version]
- Polstein, L.R.; Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-based photoactivatable transcription system. Chem. Biol. 2015, 22, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubeck, F.; Hoffmann, M.D.; Harteveld, Z.; Aschenbrenner, S.; Bietz, A.; Waldhauer, M.C.; Börner, K.; Fakhiri, J.; Schmelas, C.; Dietz, L.; et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR–Cas9. Nat. Methods 2018, 15, 924–927. [Google Scholar] [CrossRef]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Xiong, X.; Wong, S.; Charles, E.J.; Lim, W.A.; Qi, L.S. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 2016, 13, 1043–1049. [Google Scholar] [CrossRef]
- Tague, E.P.; Dotson, H.L.; Tunney, S.N.; Sloas, D.C.; Ngo, J.T. Chemogenetic control of gene expression and cell signaling with antiviral drugs. Nat. Methods 2018, 15, 519–522. [Google Scholar] [CrossRef]
- Maji, B.; Moore, C.L.; Zetsche, B.; Volz, S.E.; Zhang, F.; Shoulders, M.D.; Choudhary, A. Multidimensional chemical control of CRISPR-Cas9. Nat. Chem. Biol. 2017, 13, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Stojic, L.; Lun, A.T.L.; Mangei, J.; Mascalchi, P.; Quarantotti, V.; Barr, A.R.; Bakal, C.; Marioni, J.C.; Gergely, F.; Odom, D.T. Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res. 2018, 46, 5950–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampmann, M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine HHS Public Access. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Van Buggenhout, G.; Fryns, J.-P. Angelman syndrome (AS, MIM 105830). Eur. J. Hum. Genet. 2009, 17, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.A.; Driscoll, D.J.; Dagli, A.I. Clinical and genetic aspects of Angelman syndrome. Genet. Med. 2010, 12, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandanam, T.; Beange, H.; Robson, L.; Woolnough, H.; Buchholz, T.; Smith, A. Manifestations in institutionalised adults with Angelman syndrome due to deletion. Am. J. Med. Genet. 1997, 70, 415–420. [Google Scholar] [CrossRef]
- Malcolm, S.; Clayton-Smith, J.; Nichols, M.; Pembrey, M.E.; Armour, J.A.L.; Jeffreys, A.J.; Robb, S.; Webb, T. Uniparental paternal disomy in Angelman’s syndrome. Lancet 1991, 337, 694–697. [Google Scholar] [CrossRef]
- Buiting, K.; Groß, S.; Lich, C.; Gillessen-Kaesbach, G.; El-Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman Syndromes: A Molecular Study of 136 Patients with an Imprinting Defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, T.; Sutcliffe, J.S.; Fang, P.; Galjaard, R.-J.; Jiang, Y.; Benton, C.S.; Rommens, J.M.; Beaudet, A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997, 15, 74–77. [Google Scholar] [CrossRef]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef]
- Dittrich, B.; Robinson, W.P.; Knoblauch, H.; Buiting, K.; Schmidt, K.; Gillessen-Kaesbach, G.; Horsthemke, B. Molecular diagnosis of the Prader-Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11-13. Hum. Genet. 1992, 90, 313–315. [Google Scholar] [CrossRef]
- Kantor, B.; Kaufman, Y.; Makedonski, K.; Razin, A.; Shemer, R. Establishing the epigenetic status of the Prader–Willi/Angelman imprinting center in the gametes and embryo. Hum. Mol. Genet. 2004, 13, 2767–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rougeulle, C.; Cardoso, C.; Fontés, M.; Colleaux, L.; Lalande, M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet. 1998, 19, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, S.; van Woerden, G.M.; Bruinsma, C.F.; Mientjes, E.; Jolfaei, M.A.; Distel, B.; Kushner, S.A.; Elgersma, Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest. 2015, 125, 2069–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-S.; Allen, J.A.; Mabb, A.M.; King, I.F.; Miriyala, J.; Taylor-Blake, B.; Sciaky, N.; Dutton, J.W., Jr.; Lee, H.-M.; Chen, X.; et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Daily, J.L.; Nash, K.; Jinwal, U.; Golde, T.; Rogers, J.; Peters, M.M.; Burdine, R.D.; Dickey, C.; Banko, J.L.; Weeber, E.J. Adeno-Associated Virus-Mediated Rescue of the Cognitive Defects in a Mouse Model for Angelman Syndrome. PLoS ONE 2011, 6, e27221. [Google Scholar] [CrossRef] [Green Version]
- Bird, L.M.; Tan, W.-H.; Bacino, C.A.; Peters, S.U.; Skinner, S.A.; Anselm, I.; Barbieri-Welge, R.; Bauer-Carlin, A.; Gentile, J.K.; Glaze, D.G.; et al. A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome. Am. J. Med. Genet. A 2011, 155A, 2956–2963. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Sun, J.; Ji, A.X.; Baudry, M. Potential therapeutic approaches for Angelman syndrome. Expert Opin. Ther. Targets 2016, 20, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Miller, J.L.; Kuipers, P.J.; German, J.R.; Beaudet, A.L.; Sahoo, T.; Driscoll, D.J. Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 2012, 20, 283–290. [Google Scholar] [CrossRef]
- Wang, S.E.; Jiang, Y. Potential of Epigenetic Therapy for Prader-Willi Syndrome. Trends Pharmacol. Sci. 2019, 40, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Potter, K.J.; Burnett, L.C.; Orsso, C.E.; Inman, M.; Ryman, D.C.; Haqq, A.M. Prader–Willi-Like Phenotype Caused by an Atypical 15q11.2 Microdeletion. Genes 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazeley, P.S.; Shepelev, V.; Talebizadeh, Z.; Butler, M.G.; Fedorova, L.; Filatov, V.; Fedorov, A. snoTARGET shows that human orphan snoRNA targets locate close to alternative splice junctions. Gene 2008, 408, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef] [Green Version]
- De Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; Van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265. [Google Scholar] [CrossRef]
- Duker, A.L.; Ballif, B.C.; Bawle, E.V.; Person, R.E.; Mahadevan, S.; Alliman, S.; Thompson, R.; Traylor, R.; Bejjani, B.A.; Shaffer, L.G.; et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader–Willi syndrome. Eur. J. Hum. Genet. 2010, 18, 1196–1201. [Google Scholar] [CrossRef]
- Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte Auriol, F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; et al. Highly restricted deletion of the SNORD116 region is implicated in Prader–Willi Syndrome. Eur. J. Hum. Genet. 2015, 23, 252–255. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Christian, S.L.; Kubota, T.; Ledbetter, D.H. A 5-year-old white girl with Prader-Willi syndrome and a submicroscopic deletion of chromosome 15q11q13. Am. J. Med. Genet. 1996, 65, 137–141. [Google Scholar] [CrossRef]
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.M.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Invest. 2017, 127, 293–305. [Google Scholar] [CrossRef]
- Heksch, R.; Kamboj, M.; Anglin, K.; Obrynba, K. Review of Prader-Willi syndrome: The endocrine approach. Transl. Pediatr. 2017, 6, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Vogels, A.; De Hert, M.; Descheemaeker, M.J.; Govers, V.; Devriendt, K.; Legius, E.; Prinzie, P.; Fryns, J.P. Psychotic disorders in Prader–Willi syndrome. Am. J. Med. Genet. Part A 2004, 127A, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.A.; Ferreira, I.R.; Cintra, H.A.; Gomes, L.H.F.; Guida, L.d.C. Genotype-Phenotype Relationships and Endocrine Findings in Prader-Willi Syndrome. Front. Endocrinol. (Lausanne) 2019, 10, 864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruvinel, E.; Budinetz, T.; Germain, N.; Chamberlain, S.; Lalande, M.; Martins-Taylor, K. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader-Willi syndrome iPSCs. Hum. Mol. Genet. 2014, 23, 4674–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rauch, T.; Chen, Z.X.; Szabó, P.E.; Riggs, A.D.; Pfeifer, G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langouët, M.; Glatt-Deeley, H.R.; Chung, M.S.; Dupont-Thibert, C.M.; Mathieux, E.; Banda, E.C.; Stoddard, C.E.; Crandall, L.; Lalande, M. Zinc finger protein 274 regulates imprinted expression of transcripts in Prader-Willi syndrome neurons. Hum. Mol. Genet. 2017, 27, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lee, H.-M.; Xiong, Y.; Sciaky, N.; Hulbert, S.W.; Cao, X.; Everitt, J.I.; Jin, J.; Roth, B.L.; Jiang, Y. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader–Willi syndrome. Nat. Med. 2017, 23, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temple, I.K.; Gardner, R.J.; Robinson, D.O.; Kibirige, M.S.; Ferguson, A.W.; Baum, J.D.; Barber, J.C.K.; James, R.S.; Shield, J.P.H. Further Evidence for an Imprinted Gene for Neonatal Diabetes Localised to Chromosome 6q22–q23. Hum. Mol. Genet. 1996, 5, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Shield, J.P.; Gardner, R.J.; Wadsworth, E.J.; Whiteford, M.L.; James, R.S.; Robinson, D.O.; Baum, J.D.; Temple, I.K. Aetiopathology and genetic basis of neonatal diabetes. Arch. Dis. Child. Fetal Neonatal Ed. 1997, 76, F39–F42. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.J.; Mackay, D.J.G.; Mungall, A.J.; Polychronakos, C.; Siebert, R.; Shield, J.P.H.; Temple, I.K.; Robinson, D.O. An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet. 2000, 9, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Pivnick, E.K.; Qumsiyeh, M.B.; Tharapel, A.T.; Summitt, J.B.; Wilroy, R.S. Partial duplication of the long arm of chromosome 6: A clinically recognisable syndrome. J. Med. Genet. 1990, 27, 523–526. [Google Scholar] [CrossRef]
- Temple, I.K.; Shield, J.P.H. 6q24 transient neonatal diabetes. Rev. Endocr. Metab. Disord. 2010, 11, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, M.; Judson, H.; Okazaki, Y.; Kusakabe, M.; Muramatsu, M.; Takada, S.; Takagi, N.; Arima, T.; Wake, N.; Kamimura, K.; et al. The cell cycle control gene ZAC/PLAGL1 is imprinted—a strong candidate gene for transient neonatal diabetes. Hum. Mol. Genet. 2000, 9, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, D.; Coupe, A.M.; Shield, J.; Storr, J.; Temple, I.; Robinson, D. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum. Genet. 2002, 110, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Arima, T.; Yamasaki, K.; John, R.M.; Kato, K.; Sakumi, K.; Nakabeppu, Y.; Wake, N.; Kono, T. The human HYMAI/PLAGL1 differentially methylated region acts as an imprint control region in mice. Genomics 2006, 88, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi, A. LOT1 (ZAC1/PLAGL1) and its family members: Mechanisms and functions. J. Cell. Physiol. 2007, 210, 16–25. [Google Scholar] [CrossRef]
- Rodríguez-Henche, N.; Jamen, F.; Leroy, C.; Bockaert, J.; Brabet, P. Transcription of the mouse PAC1 receptor gene: Cell-specific expression and regulation by Zac1. Biochim. Biophys. Acta Gene Struct. Expr. 2002, 1576, 157–162. [Google Scholar]
- Arima, T.; Drewell, R.A.; Oshimura, M.; Wake, N.; Surani, M.A. A Novel Imprinted Gene, HYMAI, Is Located within an Imprinted Domain on Human Chromosome 6 Containing ZAC. Genomics 2000, 67, 248–255. [Google Scholar] [CrossRef]
- Yorifuji, T.; Higuchi, S.; Hosokawa, Y.; Kawakita, R. Chromosome 6q24-related diabetes mellitus. Clin. Pediatr. Endocrinol. 2018, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chen, X.; Shen, S.; Li, T.; Chen, L.; Hu, M.; Cao, L.; Cheng, R.; Zhao, Z.; Luo, F. Sulfonylurea in the treatment of neonatal diabetes mellitus children with heterogeneous genetic backgrounds. J. Pediatr. Endocrinol. Metab. 2015, 28, 877. [Google Scholar] [CrossRef]
- Fu, J.-L.; Wang, T.; Xiao, X.-H. Relapsed 6q24-related transient neonatal diabetes mellitus successfully treated with sulfonylurea. Chin. Med. J. (Engl.) 2019, 132, 846–848. [Google Scholar] [CrossRef]
- Abu-Amero, S.; Monk, D.; Frost, J.; Preece, M.; Stanier, P.; Moore, G.E. The genetic aetiology of Silver-Russell syndrome. J. Med. Genet. 2008, 45, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Gicquel, C.; Rossignol, S.; Cabrol, S.; Houang, M.; Steunou, V.; Barbu, V.; Danton, F.; Thibaud, N.; Merrer, M.L.; Burglen, L.; et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet. 2005, 37, 1003–1007. [Google Scholar] [CrossRef]
- Netchine, I.; Rossignol, S.; Dufourg, M.-N.; Azzi, S.; Rousseau, A.; Perin, L.; Houang, M.; Steunou, V.; Esteva, B.; Thibaud, N.; et al. 11p15 Imprinting Center Region 1 Loss of Methylation Is a Common and Specific Cause of Typical Russell-Silver Syndrome: Clinical Scoring System and Epigenetic-Phenotypic Correlations. J. Clin. Endocrinol. Metab. 2007, 92, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Rossignol, S.; Steunou, V.; Sas, T.; Thibaud, N.; Danton, F.; Le Jule, M.; Heinrichs, C.; Cabrol, S.; Gicquel, C.; et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 2009, 18, 4724–4733. [Google Scholar] [CrossRef] [Green Version]
- Gaston, V.; Le Bouc, Y.; Soupre, V.; Burglen, L.; Donadieu, J.; Oro, H.; Audry, G.; Vazquez, M.-P.; Gicquel, C. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith–Wiedemann syndrome. Eur. J. Hum. Genet. 2001, 9, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Pianka, M.A.; McIntosh, A.T.; Patel, S.D.; Bakhshi, P.R.; Jung, M. Close yet so far away: A look into the management strategies of genetic imprinting disorders. Am. J. Stem Cells 2018, 7, 72–81. [Google Scholar]
- Eggermann, T.; Begemann, M.; Binder, G.; Spengler, S. Silver-Russell syndrome: Genetic basis and molecular genetic testing. Orphanet J. Rare Dis. 2010, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Saal, H.M.; Harbison, M.D.; Netchine, I. Silver-Russell Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Han, L.; Szabó, P.E.; Mann, J.R. Postnatal Survival of Mice with Maternal Duplication of Distal Chromosome 7 Induced by a Igf2/H19 Imprinting Control Region Lacking Insulator Function. PLoS Genet. 2010, 6, e1000803. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Imprinting Disorder | Chromosome/Gene | Mutation/Epimutation | Frequency |
|---|---|---|---|
| Angelman Syndrome | 15q11.2-13q | Maternal deletion | 1:12,000/1:20,000 |
| UPD(15)Pat | |||
| Methylation defects | |||
| Ube3a | Point mutations | ||
| Prader-Willi Syndrome | 15q11.2-13q | Paternal deletion | 1:10,000/1:25,000 |
| UPD(15)Mat | |||
| Methylation defects | |||
| Transient Neonatal Diabetes Mellitus | 6q24, PLAGL1/HYMA1 | UPD(6)Pat | 1:400,000 |
| Paternal duplication | |||
| Methylation defects | |||
| Silver-Russell Syndrome | 7 | UPD(7)Mat | 1:75,000/1:100,000 |
| 11p15 | UPD(11p15)Mat | ||
| Maternal duplication | |||
| IGF2/H19 | Paternal hypomethylation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993. https://doi.org/10.3390/cells9040993
Syding LA, Nickl P, Kasparek P, Sedlacek R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells. 2020; 9(4):993. https://doi.org/10.3390/cells9040993
Chicago/Turabian StyleSyding, Linn Amanda, Petr Nickl, Petr Kasparek, and Radislav Sedlacek. 2020. "CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review" Cells 9, no. 4: 993. https://doi.org/10.3390/cells9040993
APA StyleSyding, L. A., Nickl, P., Kasparek, P., & Sedlacek, R. (2020). CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells, 9(4), 993. https://doi.org/10.3390/cells9040993