Lysosomal Biology and Function: Modern View of Cellular Debris Bin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
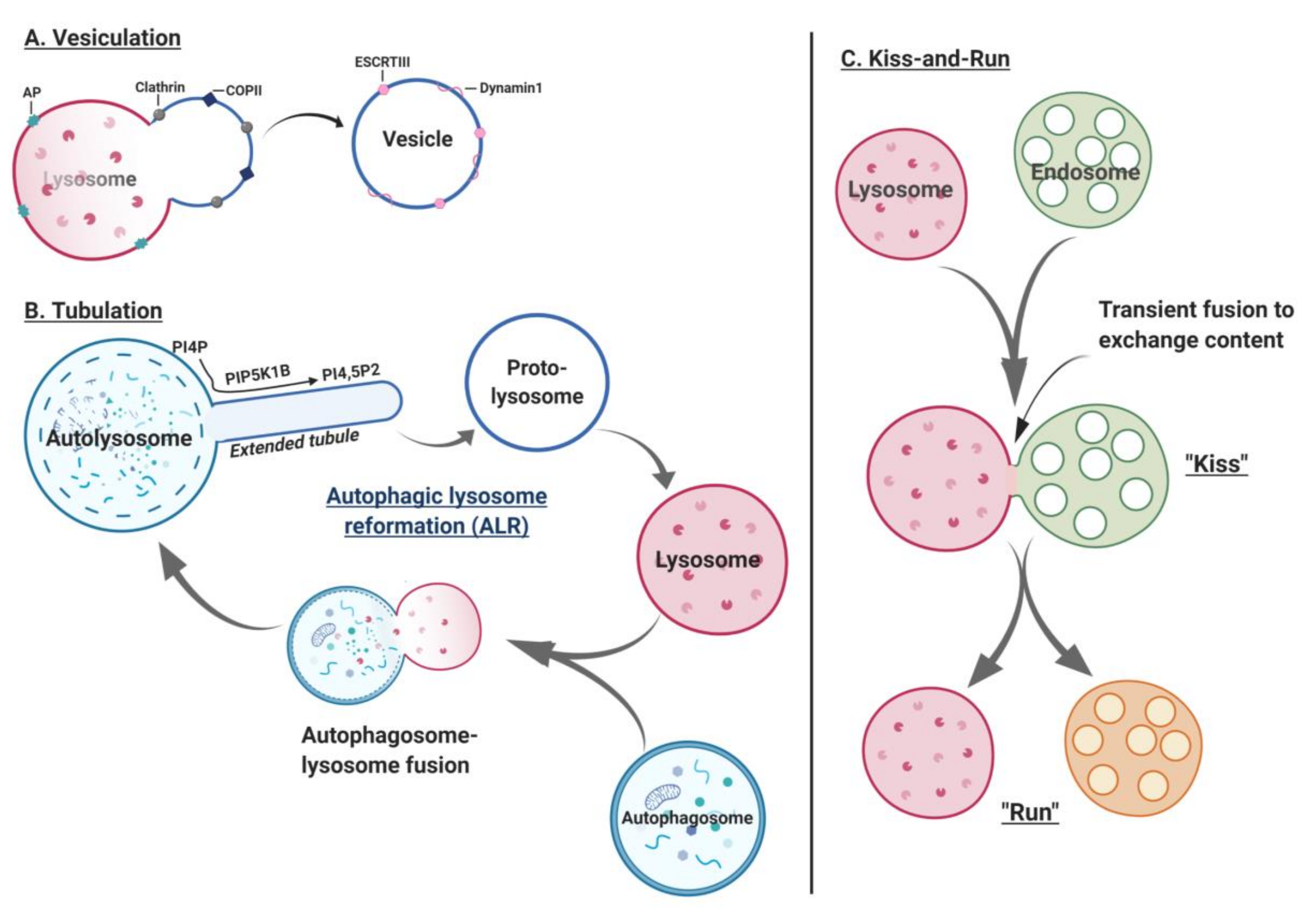{kind=link}
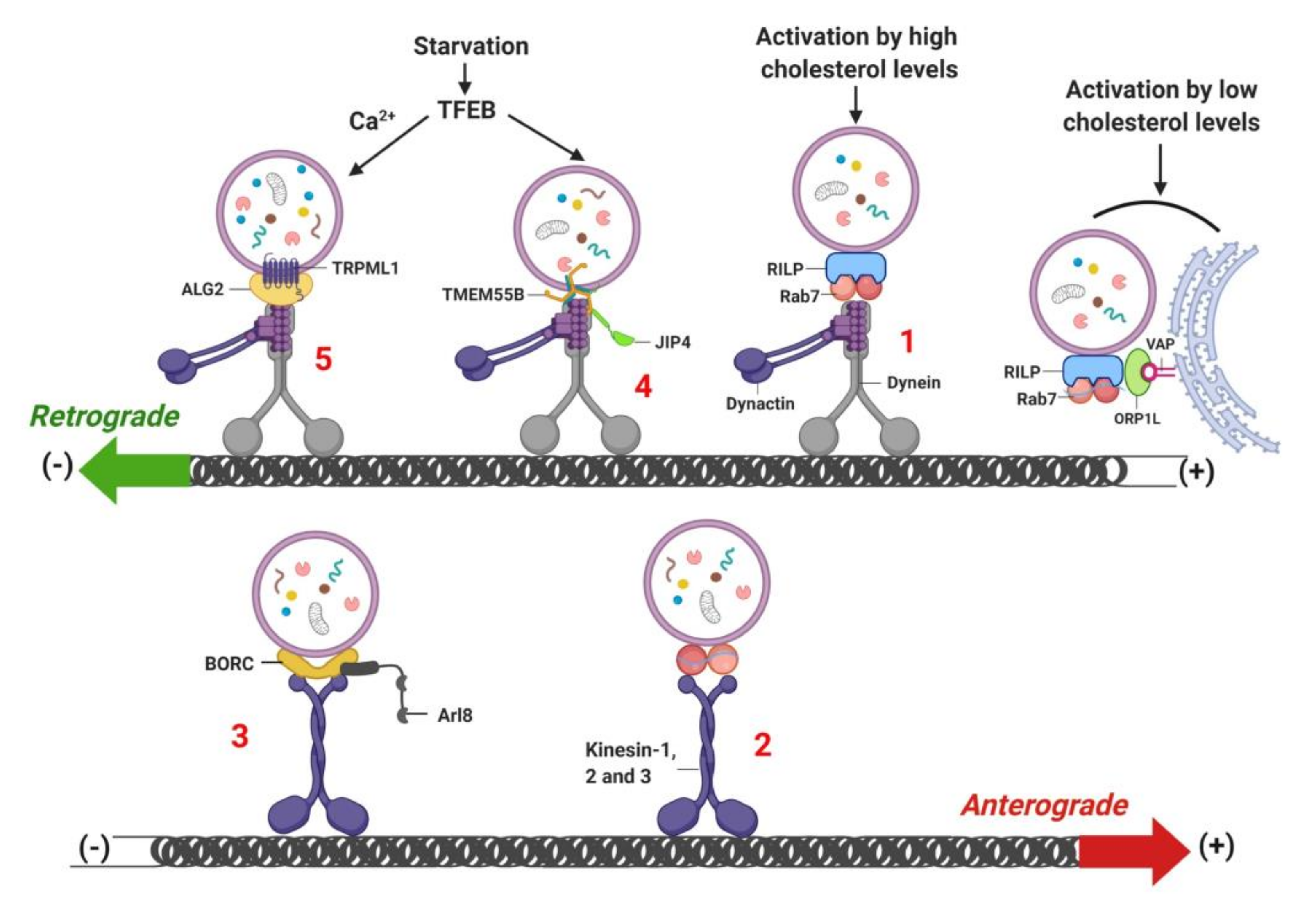{kind=link}
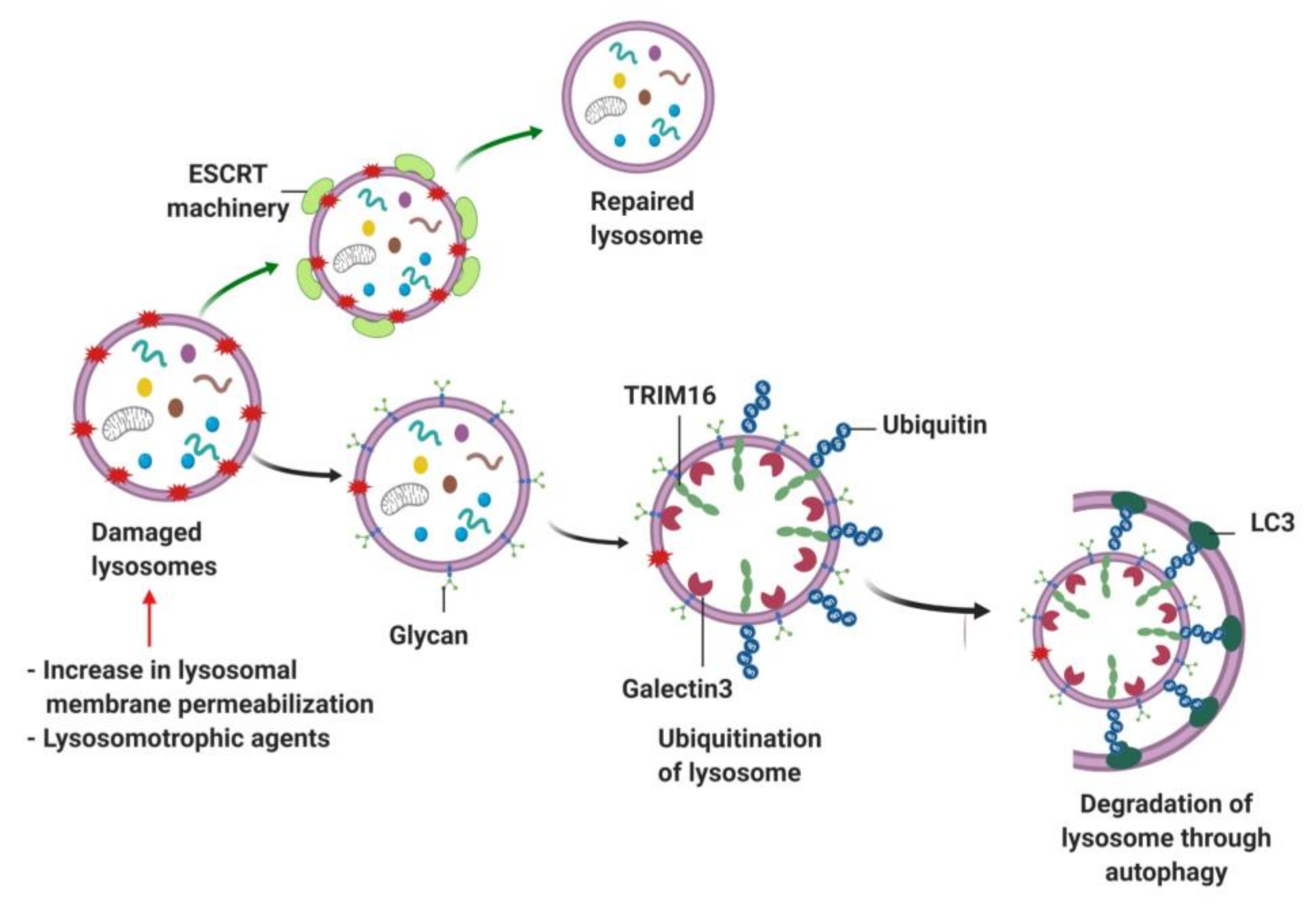{kind=link}
Abstract
:1. Introduction
2. Lysosome Biogenesis
2.1. Sorting
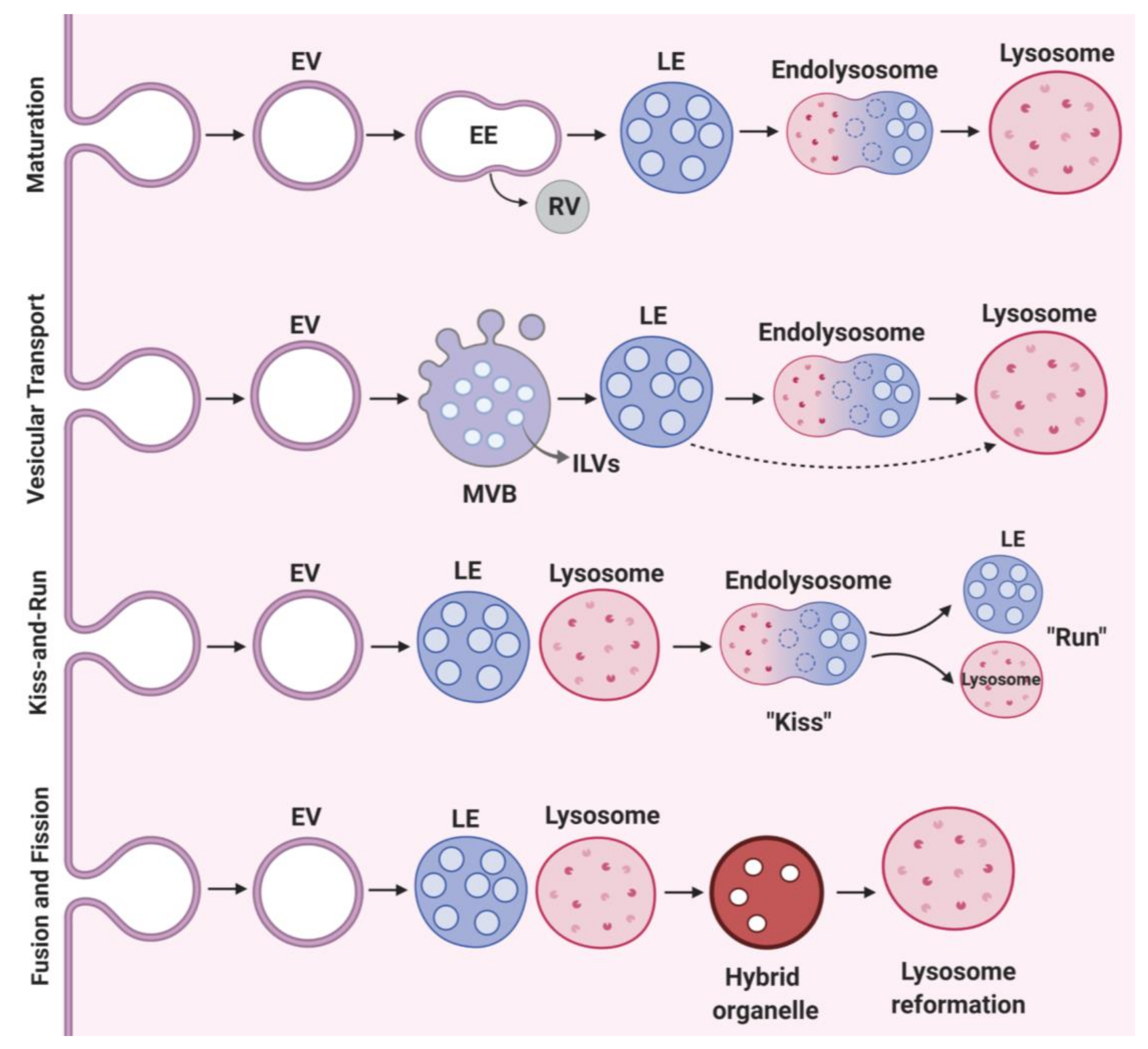
2.2. Vesicular Transport and Maturation
3. Lysosomal Enzymes
3.1. Cathepsin Proteases
3.2. Lysosomal Acid Lipases (LAL)
3.3. Sulfatases
3.4. Nucleases
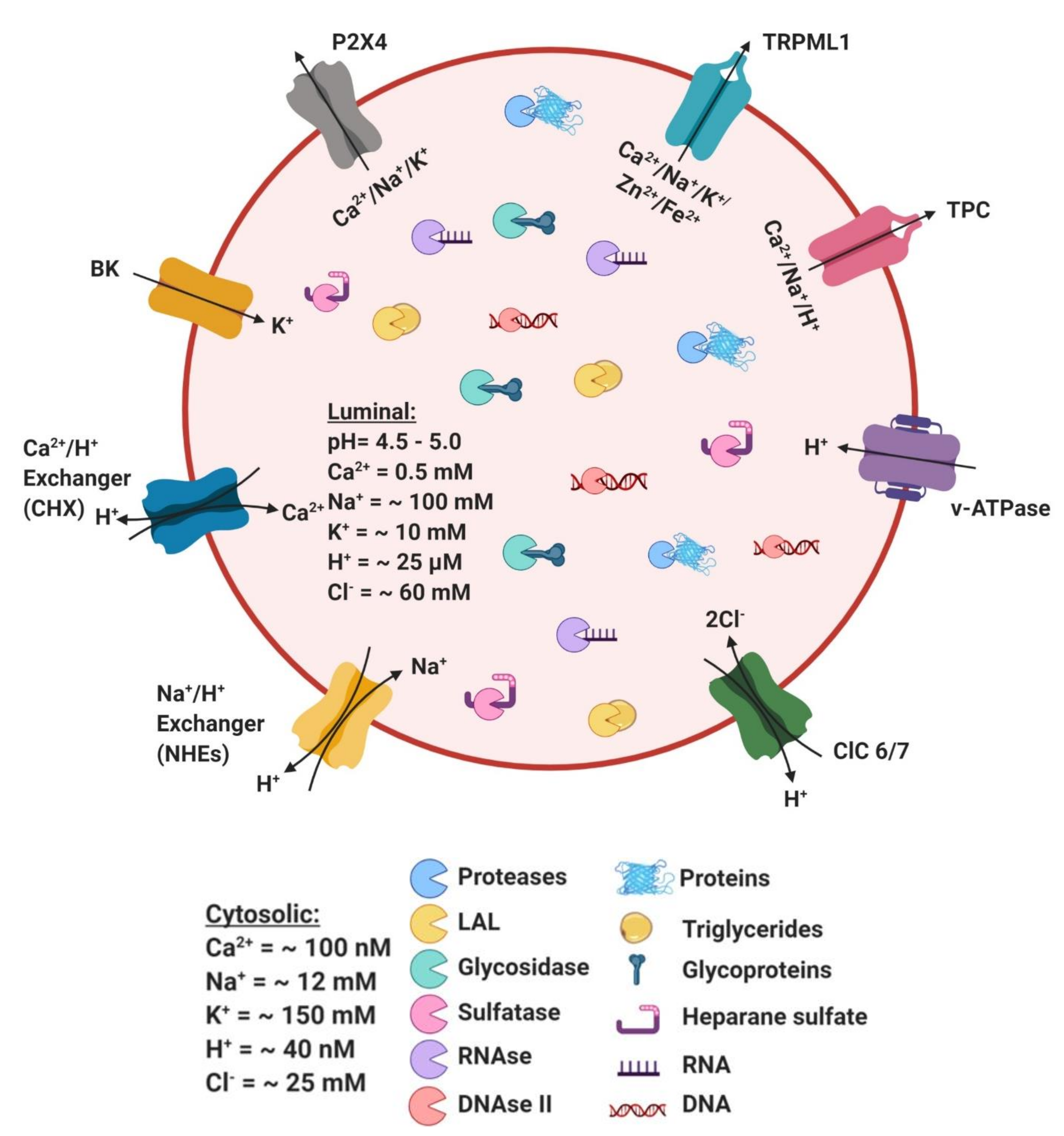
4. Ionic Balance in Lysosome Function
4.1. Ca2+
4.2. H+
4.3. Na+/K+
4.4. Cl−
4.5. Fe2+ and Zn2+
5. Role of Ion Channels in Lysosomal Function
5.1. Transient Receptor Potential Cation Channel, Mucolipin Subfamily (TRPML)
5.2. Two-Pore Channels (TPC)
5.3. P2X4
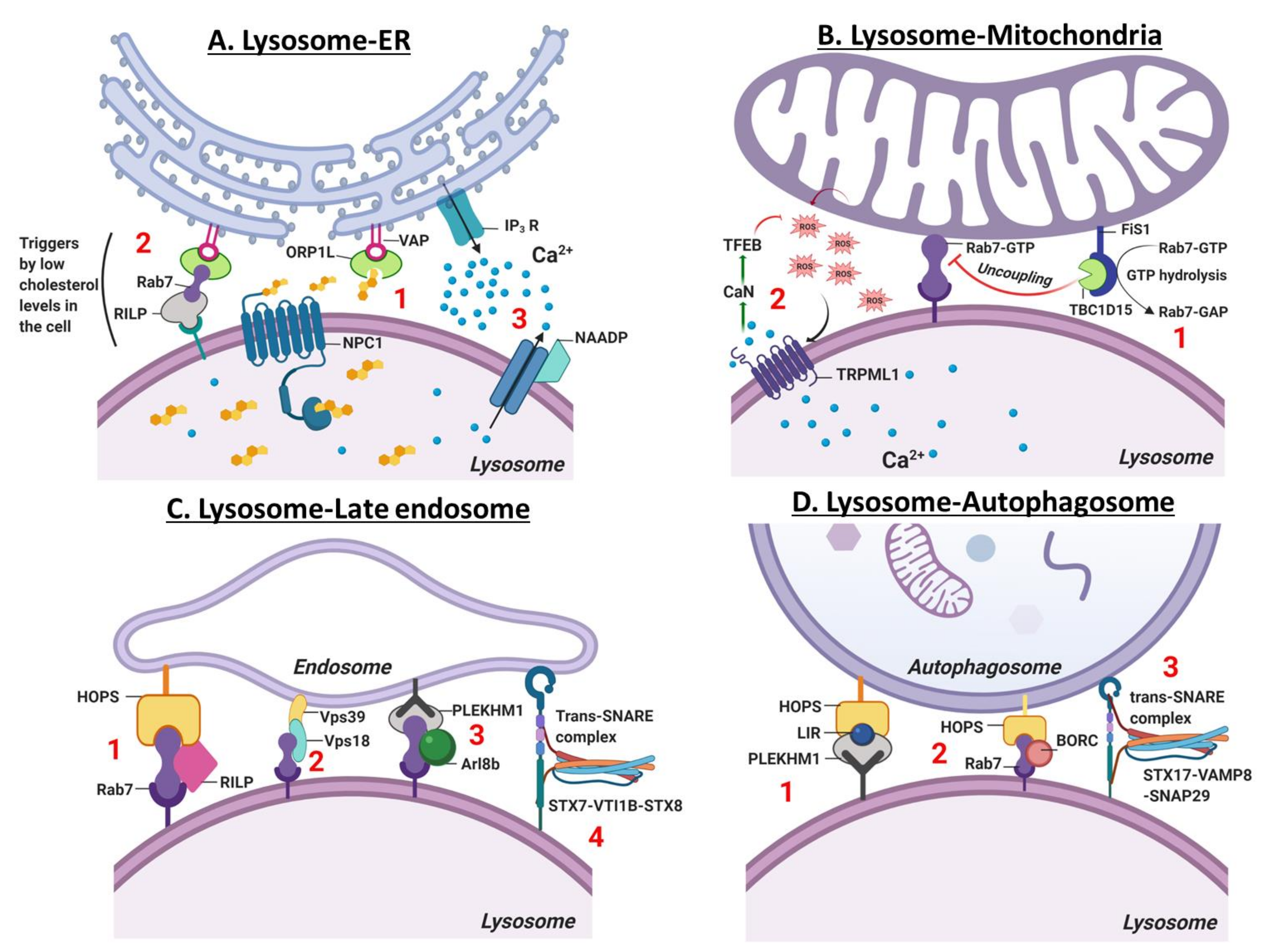
6. Contact Site Between the Lysosome and other Organelles
6.1. Lysosome-ER Contact Site
6.2. Lysosome-Mitochondria Contact Site
7. Lysosome Fusion and Fission Events
7.1. Late Endosome-Lysosome-Autophagosome Fusion
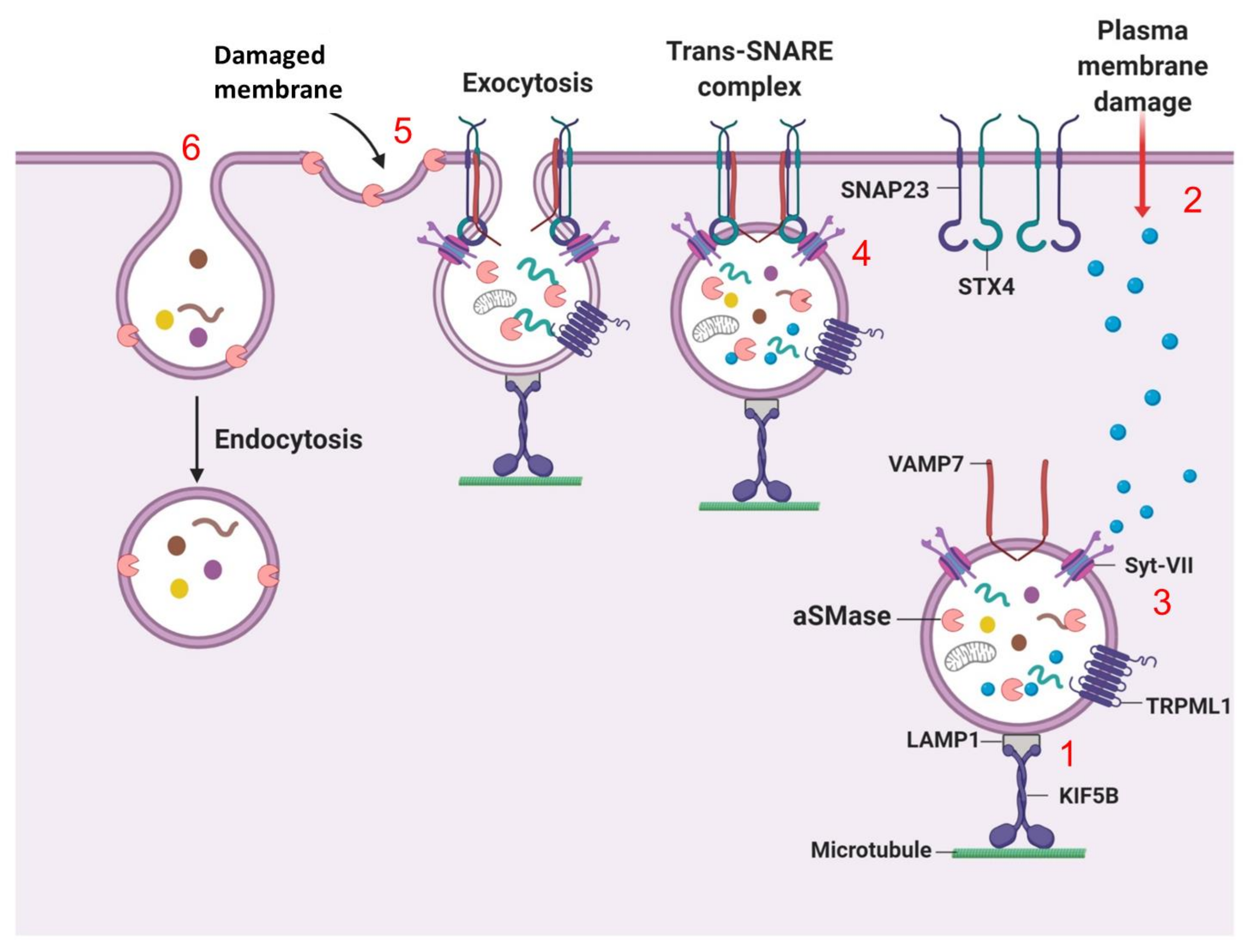
7.2. Lysosomal Exocytosis (Fusion of Lysosomes with the Plasma Membrane)
7.3. Lysosome Fission
8. Lysosome Localization and Movement
9. Lysosome-Dependent Protein Degradation
9.1. Macroautophagy
9.2. Lipophagy, Mitophagy and Lysophagy
9.3. Chaperone-Mediated Autophagy (CMA)
9.4. Microautophagy
10. Lysosome Dysfunction in Diseases
10.1. Lysosomal Storage Disorder (LSDs)
10.2. Neurodegenerative Diseases
10.3. Cardiovascular Diseases
11. Conclusion and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Bainton, D.F. The discovery of lysosomes. J. Cell Biol. 1981, 91, 66s–76s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, P.R.; Luzio, J.P. Delivery of endocytosed membrane proteins to the lysosome. Biochim. Biophys. Acta 2009, 1793, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.B.; Dammer, E.B.; Ren, R.J.; Wang, G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Jegga, A.G.; Schneider, L.; Ouyang, X.; Zhang, J. Systems biology of the autophagy-lysosomal pathway. Autophagy 2011, 7, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Herb, M.; Gluschko, A.; Schramm, M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Gray, M.A.; Choy, C.H.; Dayam, R.M.; Ospina-Escobar, E.; Somerville, A.; Xiao, X.; Ferguson, S.M.; Botelho, R.J. Phagocytosis Enhances Lysosomal and Bactericidal Properties by Activating the Transcription Factor TFEB. Curr. Biol. CB 2016, 26, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Schwake, M.; Schroder, B.; Saftig, P. Lysosomal membrane proteins and their central role in physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, M.X. Regulation of lysosomal ion homeostasis by channels and transporters. Sci. China Life Sci. 2016, 59, 777–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Biol. 2019, 29, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Castro-Gomes, T. Lysosomes and plasma membrane repair. Curr. Top. Membr. 2019, 84, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, M.E.G.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal size matters. Traffic 2020, 21, 60–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193. [Google Scholar] [CrossRef] [Green Version]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Gray, S.R.; Gratian, M.J.; Piper, R.C.; Bright, N.A. Membrane traffic to and from lysosomes. Biochem. Soc. Symp. 2005, 77–86. [Google Scholar] [CrossRef]
- Woychik, N.A.; Cardelli, J.A.; Dimond, R.L. A conformationally altered precursor to the lysosomal enzyme alpha-mannosidase accumulates in the endoplasmic reticulum in a mutant strain of Dictyostelium discoideum. J. Biol. Chem. 1986, 261, 9595–9602. [Google Scholar]
- Doray, B.; Ghosh, P.; Griffith, J.; Geuze, H.J.; Kornfeld, S. Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science 2002, 297, 1700–1703. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.N. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 2004, 165, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Sachse, M.; Urbe, S.; Oorschot, V.; Strous, G.J.; Klumperman, J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell 2002, 13, 1313–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubakar, Y.S.; Zheng, W.; Olsson, S.; Zhou, J. Updated Insight into the Physiological and Pathological Roles of the Retromer Complex. Int. J. Mol. Sci. 2017, 18, 1601. [Google Scholar] [CrossRef] [Green Version]
- Wandinger-Ness, A.; Zerial, M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Bowman, S.L.; Bi-Karchin, J.; Le, L.; Marks, M.S. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic 2019, 20, 404–435. [Google Scholar] [CrossRef] [Green Version]
- Ivan, V.; Martinez-Sanchez, E.; Sima, L.E.; Oorschot, V.; Klumperman, J.; Petrescu, S.M.; van der Sluijs, P. AP-3 and Rabip4’ coordinately regulate spatial distribution of lysosomes. PLoS ONE 2012, 7, e48142. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: the peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogalingam, G.; Pendergast, A.M. Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J. Biol. Chem. 2008, 283, 35941–35953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgens, E.; Lutgens, S.P.; Faber, B.C.; Heeneman, S.; Gijbels, M.M.; de Winther, M.P.; Frederik, P.; van der Made, I.; Daugherty, A.; Sijbers, A.M.; et al. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation 2006, 113, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamoto, S.; Sukhova, G.K.; Sun, J.; Yang, M.; Libby, P.; Love, V.; Duramad, P.; Sun, C.; Zhang, Y.; Yang, X.; et al. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation 2007, 115, 2065–2075. [Google Scholar] [CrossRef]
- Lutgens, S.P.; Cleutjens, K.B.; Daemen, M.J.; Heeneman, S. Cathepsin cysteine proteases in cardiovascular disease. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3029–3041. [Google Scholar] [CrossRef]
- Hakala, J.K.; Oksjoki, R.; Laine, P.; Du, H.; Grabowski, G.A.; Kovanen, P.T.; Pentikainen, M.O. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.W.; Shi, G.P.; Kuzuya, M.; Sasaki, T.; Okumura, K.; Murohara, T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation 2012, 125, 1551–1562. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.F.; Herrington, D.M. The function of cathepsins B, D, and X in atherosclerosis. Am. J. Cardiovasc. Dis. 2016, 6, 163–170. [Google Scholar]
- Helske, S.; Syvaranta, S.; Kupari, M.; Lappalainen, J.; Laine, M.; Lommi, J.; Turto, H.; Mayranpaa, M.; Werkkala, K.; Kovanen, P.T.; et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur. Heart J. 2006, 27, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Lindstedt, K.A.; Mayranpaa, M.I.; Kovanen, P.T. Mast cells in vulnerable atherosclerotic plaques--a view to a kill. J. Cell. Mol. Med. 2007, 11, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Felbor, U.; Kessler, B.; Mothes, W.; Goebel, H.H.; Ploegh, H.L.; Bronson, R.T.; Olsen, B.R. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc. Natl. Acad. Sci. USA 2002, 99, 7883–7888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Authier, F.; Metioui, M.; Fabrega, S.; Kouach, M.; Briand, G. Endosomal proteolysis of internalized insulin at the C-terminal region of the B chain by cathepsin D. J. Biol. Chem. 2002, 277, 9437–9446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlanetto, R.W. Receptor-mediated endocytosis and lysosomal processing of insulin-like growth factor I by mitogenically responsive cells. Endocrinology 1988, 122, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Authier, F.; Kouach, M.; Briand, G. Endosomal proteolysis of insulin-like growth factor-I at its C-terminal D-domain by cathepsin B. FEBS Lett. 2005, 579, 4309–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwad, O.; Kubler, B.; Roth, W.; Scharf, J.G.; Saftig, P.; Peters, C.; Braulke, T. Decreased intracellular degradation of insulin-like growth factor binding protein-3 in cathepsin L-deficient fibroblasts. FEBS Lett. 2002, 510, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Ohya, Y.; Suzuki, K. Atg4 plays an important role in efficient expansion of autophagic isolation membranes by cleaving lipidated Atg8 in Saccharomyces cerevisiae. PLoS ONE 2017, 12, e0181047. [Google Scholar] [CrossRef] [Green Version]
- Kaminskyy, V.; Zhivotovsky, B. Proteases in autophagy. Biochim. Biophys. Acta 2012, 1824, 44–50. [Google Scholar] [CrossRef]
- Maruyama, T.; Noda, N.N. Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. J. Antibiot. 2017. [Google Scholar] [CrossRef] [Green Version]
- Yanagawa, M.; Tsukuba, T.; Nishioku, T.; Okamoto, Y.; Okamoto, K.; Takii, R.; Terada, Y.; Nakayama, K.I.; Kadowaki, T.; Yamamoto, K. Cathepsin E deficiency induces a novel form of lysosomal storage disorder showing the accumulation of lysosomal membrane sialoglycoproteins and the elevation of lysosomal pH in macrophages. J. Biol. Chem. 2007, 282, 1851–1862. [Google Scholar] [CrossRef] [Green Version]
- Dubland, J.A.; Francis, G.A. Lysosomal acid lipase: at the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yuan, X.; Li, N.; Dewey, W.L.; Li, P.L.; Zhang, F. Lysosomal cholesterol accumulation in macrophages leading to coronary atherosclerosis in CD38(-/-) mice. J. Cell. Mol. Med. 2016, 20, 1001–1013. [Google Scholar] [CrossRef]
- Jerome, W.G. Lysosomes, cholesterol and atherosclerosis. Clin. Lipidol. 2010, 5, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Coltoff-Schiller, B.; Goldfischer, S.; Wolinsky, H.; Factor, S.M. Lipid accumulation in human aortic smooth muscle cell lysosomes. Am. J. Pathol. 1976, 83, 39–44. [Google Scholar] [PubMed]
- Jerome, W.G.; Yancey, P.G. The role of microscopy in understanding atherosclerotic lysosomal lipid metabolism. Microsc. Microanal. 2003, 9, 54–67. [Google Scholar] [CrossRef]
- Goldfischer, S.; Schiller, B.; Wolinsky, H. Lipid accumulation in smooth muscle cell lysosomes im primate atherosclerosis. Am. J. Pathol. 1975, 78, 497–504. [Google Scholar]
- Shio, H.; Farquhar, M.G.; de Duve, C. Lysosomes of the arterial wall. IV. Cytochemical localization of acid phosphatase and catalase in smooth muscle cells and foam cells from rabbit atheromatous aorta. Am. J. Pathol. 1974, 76, 1–16. [Google Scholar]
- Peters, T.J.; Muller, M.; De Duve, C. Lysosomes of the arterial wall. I. Isolation and subcellular fractionation of cells from normal rabbit aorta. J. Exp. Med. 1972, 136, 1117–1139. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.A.; Liu, B.; Nazarenko, I.; Martis, S.; Kozlitina, J.; Yang, Y.; Ramirez, C.; Kasai, Y.; Hyatt, T.; Peter, I.; et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology 2013, 58, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Stitziel, N.O.; Fouchier, S.W.; Sjouke, B.; Peloso, G.M.; Moscoso, A.M.; Auer, P.L.; Goel, A.; Gigante, B.; Barnes, T.A.; Melander, O.; et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2909–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Schiavi, S.; Wan, N.; Levine, M.; Witte, D.P.; Grabowski, G.A. Reduction of atherosclerotic plaques by lysosomal acid lipase supplementation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejko, J.J. Managing Cardiovascular Risk in Lysosomal Acid Lipase Deficiency. Am. J. Cardiovasc. Drugs 2017, 17, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Donaldson, E.; Sharma, R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl. Clin. Genet. 2016, 9, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, P.; Du, H.; Wilkes, D.S.; Yan, C. Critical roles of lysosomal acid lipase in T cell development and function. Am. J. Pathol. 2009, 174, 944–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, M.H.; Watts, G.F. Increased hepatic secretion of very-low-density lipoprotein apolipoprotein B-100 in cholesteryl ester storage disease. Clin. Chem. 1995, 41, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar]
- Hanson, S.R.; Best, M.D.; Wong, C.H. Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. 2004, 43, 5736–5763. [Google Scholar] [CrossRef]
- Sardiello, M.; Annunziata, I.; Roma, G.; Ballabio, A. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum. Mol. Genet. 2005, 14, 3203–3217. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Selmer, T.; Ingendoh, A.; von Figura, K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995, 82, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Wada, K.; Kabuta, T. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J. Biochem. 2017, 161, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, Y.; Furuta, A.; Kikuchi, H.; Aizawa, S.; Hatanaka, Y.; Konya, C.; Uchida, K.; Yoshimura, A.; Tamai, Y.; Wada, K.; et al. Discovery of a novel type of autophagy targeting RNA. Autophagy 2013, 9, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, Y.; Kikuchi, H.; Aizawa, S.; Furuta, A.; Hatanaka, Y.; Konya, C.; Uchida, K.; Wada, K.; Kabuta, T. Direct uptake and degradation of DNA by lysosomes. Autophagy 2013, 9, 1167–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campomenosi, P.; Salis, S.; Lindqvist, C.; Mariani, D.; Nordstrom, T.; Acquati, F.; Taramelli, R. Characterization of RNASET2, the first human member of the Rh/T2/S family of glycoproteins. Arch. Biochem. Biophys. 2006, 449, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.J.; Aguilera, R.J. DNase II: genes, enzymes and function. Gene 2003, 322, 1–15. [Google Scholar] [CrossRef]
- Luhtala, N.; Parker, R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, K.W.; Fink, G.R. Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J. Cell Biol. 1994, 124, 351–363. [Google Scholar] [CrossRef]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca2+ homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef]
- Melchionda, M.; Pittman, J.K.; Mayor, R.; Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 2016, 212, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Mayer, A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature 1998, 396, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The role of intraorganellar Ca2+ in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J. Cell Biol. 2000, 149, 1053–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; Ostrowski, P.; Jaumouille, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, J.; Grinstein, S. Na+/H+ exchangers of mammalian cells. J. Biol. Chem. 1997, 272, 22373–22376. [Google Scholar] [CrossRef] [Green Version]
- Stauber, T.; Jentsch, T.J. Chloride in vesicular trafficking and function. Annu. Rev. Physiol. 2013, 75, 453–477. [Google Scholar] [CrossRef] [Green Version]
- Graves, A.R.; Curran, P.K.; Smith, C.L.; Mindell, J.A. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 2008, 453, 788–792. [Google Scholar] [CrossRef]
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.C.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poet, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.V.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H.; et al. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol. Cell 2020, 77, 645–655. [Google Scholar] [CrossRef]
- Kidane, T.Z.; Sauble, E.; Linder, M.C. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol Cell Physiol. 2006, 291, C445–C455. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Gitschier, J. A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat. Genet. 1997, 17, 292–297. [Google Scholar] [CrossRef]
- McCormick, N.H.; Kelleher, S.L. ZnT4 provides zinc to zinc-dependent proteins in the trans-Golgi network critical for cell function and Zn export in mammary epithelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C291–C297. [Google Scholar] [CrossRef] [Green Version]
- Eichelsdoerfer, J.L.; Evans, J.A.; Slaugenhaupt, S.A.; Cuajungco, M.P. Zinc dyshomeostasis is linked with the loss of mucolipidosis IV-associated TRPML1 ion channel. J. Biol. Chem. 2010, 285, 34304–34308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Hua, Y.; Vergarajauregui, S.; Diab, H.I.; Puertollano, R. Novel Role of TRPML2 in the Regulation of the Innate Immune Response. J. Immunol. 2015, 195, 4922–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatachalam, K.; Hofmann, T.; Montell, C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J. Biol. Chem. 2006, 281, 17517–17527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Long, A.A.; Elsaesser, R.; Nikolaeva, D.; Broadie, K.; Montell, C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 2008, 135, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Samie, M.A.; Xu, H. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 2014, 55, 995–1009. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [Green Version]
- Karacsonyi, C.; Miguel, A.S.; Puertollano, R. Mucolipin-2 localizes to the Arf6-associated pathway and regulates recycling of GPI-APs. Traffic 2007, 8, 1404–1414. [Google Scholar] [CrossRef] [Green Version]
- Cuajungco, M.P.; Silva, J.; Habibi, A.; Valadez, J.A. The mucolipin-2 (TRPML2) ion channel: a tissue-specific protein crucial to normal cell function. Pflug. Arch. Eur. J. Physiol. 2016, 468, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Remis, N.N.; Wiwatpanit, T.; Castiglioni, A.J.; Flores, E.N.; Cantu, J.A.; Garcia-Anoveros, J. Mucolipin co-deficiency causes accelerated endolysosomal vacuolation of enterocytes and failure-to-thrive from birth to weaning. PLoS Genet. 2014, 10, e1004833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, T.; Cai, X.; Brailoiu, G.C.; Abood, M.E.; Brailoiu, E.; Patel, S. Two-pore channels provide insight into the evolution of voltage-gated Ca2+ and Na+ channels. Sci. Signal. 2014, 7, ra109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. J. Biol. Chem. 2015, 290, 3377–3389. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.J.; Hirata, H.; Fimia, G.M.; do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef] [Green Version]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Ruas, M.; Davis, L.C.; Chen, C.C.; Morgan, A.J.; Chuang, K.T.; Walseth, T.F.; Grimm, C.; Garnham, C.; Powell, T.; Platt, N.; et al. Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J. 2015, 34, 1743–1758. [Google Scholar] [CrossRef]
- Capel, R.A.; Bolton, E.L.; Lin, W.K.; Aston, D.; Wang, Y.; Liu, W.; Wang, X.; Burton, R.A.; Bloor-Young, D.; Shade, K.T.; et al. Two-pore Channels (TPC2s) and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) at Lysosomal-Sarcoplasmic Reticular Junctions Contribute to Acute and Chronic beta-Adrenoceptor Signaling in the Heart. J. Biol. Chem. 2015, 290, 30087–30098. [Google Scholar] [CrossRef] [Green Version]
- Bazowska, G.; Jendryczko, A.; Dudkiewicz, J. Evaluation of amniotic fluid elastolytic activity: can it be a method of fetal lung maturity assessment? A comparison with Gluck’s L/S test. Acta Genet. Med. Gemellol. (Roma) 1994, 43, 185–192. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Paramasivam, A.; Yu, J.C.; Murrell-Lagnado, R.D. Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J. Cell Sci. 2007, 120, 3838–3849. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrell-Lagnado, R.; Zhu, M.X.; Dong, X.P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 2015, 209, 879–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [PubMed]
- Huang, P.; Zou, Y.; Zhong, X.Z.; Cao, Q.; Zhao, K.; Zhu, M.X.; Murrell-Lagnado, R.; Dong, X.P. P2X4 forms functional ATP-activated cation channels on lysosomal membranes regulated by luminal pH. J. Biol. Chem. 2014, 289, 17658–17667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Dibenedetto, J.R.; West, M.; Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol. Biol. Cell 2013, 24, 1030–1040. [Google Scholar] [CrossRef]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Kumar, J.; Ferguson, C.; Schulz, T.A.; Ong, Y.S.; Hong, W.; Prinz, W.A.; Parton, R.G.; Brown, A.J.; Yang, H. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J. Cell Biol. 2011, 192, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Jiang, L.; Yang, H.; Song, B.L. Routes and mechanisms of post-endosomal cholesterol trafficking: A story that never ends. Traffic 2017, 18, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol. 2017, 216, 4217–4233. [Google Scholar] [CrossRef] [Green Version]
- Ko, D.C.; Gordon, M.D.; Jin, J.Y.; Scott, M.P. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol. Biol. Cell 2001, 12, 601–614. [Google Scholar] [CrossRef]
- Churchill, G.C.; Galione, A. NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J. 2001, 20, 2666–2671. [Google Scholar] [CrossRef]
- Kinnear, N.P.; Boittin, F.X.; Thomas, J.M.; Galione, A.; Evans, A.M. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 2004, 279, 54319–54326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, X.; Schieder, M.; Cuny, H.; Fenske, S.; Gruner, C.; Rotzer, K.; Griesbeck, O.; Harz, H.; Biel, M.; Wahl-Schott, C. The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflug. Arch. Eur. J. Physiol. 2009, 458, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Galione, A. A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Penny, C.J.; Kilpatrick, B.S.; Han, J.M.; Sneyd, J.; Patel, S. A computational model of lysosome-ER Ca2+ microdomains. J. Cell Sci. 2014, 127, 2934–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca(2)(+) signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Jin, C.; Shao, X.; Guan, R.; Tian, Z.; Wang, C.; Liu, F.; Ling, P.; Guan, J.L.; Ji, L.; et al. Super-Resolution Tracking of Mitochondrial Dynamics with An Iridium(III) Luminophore. Small 2018, 14, e1802166. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Mosquera, L.; Diogo, C.V.; Yambire, K.F.; Santos, G.L.; Luna Sanchez, M.; Benit, P.; Rustin, P.; Lopez, L.C.; Milosevic, I.; Raimundo, N. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci Rep. 2017, 7, 45076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullock, B.M.; Branch, W.J.; van Schaik, M.; Gilbert, L.K.; Luzio, J.P. Reconstitution of an endosome-lysosome interaction in a cell-free system. J. Cell Biol. 1989, 108, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. CB 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Marwaha, R.; Arya, S.B.; Jagga, D.; Kaur, H.; Tuli, A.; Sharma, M. The Rab7 effector PLEKHM1 binds Arl8b to promote cargo traffic to lysosomes. J. Cell Biol. 2017, 216, 1051–1070. [Google Scholar] [CrossRef] [PubMed]
- Caplan, S.; Hartnell, L.M.; Aguilar, R.C.; Naslavsky, N.; Bonifacino, J.S. Human Vam6p promotes lysosome clustering and fusion in vivo. J. Cell Biol. 2001, 154, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Poupon, V.; Stewart, A.; Gray, S.R.; Piper, R.C.; Luzio, J.P. The role of mVps18p in clustering, fusion, and intracellular localization of late endocytic organelles. Mol. Biol. Cell 2003, 14, 4015–4027. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.C.; Winistorfer, S.C.; Poupon, V.; Luzio, J.P.; Piper, R.C. Mammalian late vacuole protein sorting orthologues participate in early endosomal fusion and interact with the cytoskeleton. Mol. Biol. Cell 2004, 15, 1197–1210. [Google Scholar] [CrossRef]
- Khatter, D.; Raina, V.B.; Dwivedi, D.; Sindhwani, A.; Bahl, S.; Sharma, M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci. 2015, 128, 1746–1761. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Sharma, M.; Ung, C.; Tuli, A.; Barral, D.C.; Hava, D.L.; Veerapen, N.; Besra, G.S.; Hacohen, N.; Brenner, M.B. Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity 2011, 35, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.; Bredahl, B.; Ostermann, K.M. What consequences do the EEC directives have for our work environment? Sygeplejersken 1981, 81, 12–13. [Google Scholar]
- Kuo, W.; Herrick, D.Z.; Ellena, J.F.; Cafiso, D.S. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: Two modes of C2B binding. J. Mol. Biol. 2009, 387, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonin, W.; Holroyd, C.; Fasshauer, D.; Pabst, S.; Von Mollard, G.F.; Jahn, R. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. EMBO J. 2000, 19, 6453–6464. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parton, R.G.; Dotti, C.G.; Bacallao, R.; Kurtz, I.; Simons, K.; Prydz, K. pH-induced microtubule-dependent redistribution of late endosomes in neuronal and epithelial cells. J. Cell Biol. 1991, 113, 261–274. [Google Scholar] [CrossRef] [Green Version]
- McEwan, D.G.; Dikic, I. PLEKHM1: Adapting to life at the lysosome. Autophagy 2015, 11, 720–722. [Google Scholar] [CrossRef] [Green Version]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; de Stegmann, D.M.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Guardia, C.M.; Pu, J.; Chen, Y.; Bonifacino, J.S. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 2017, 13, 1648–1663. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W. Lysosomes and the plasma membrane: trypanosomes reveal a secret relationship. J. Cell Biol. 2002, 158, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, C.M.; Groth-Pedersen, L.; Hoyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jaattela, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Kobayashi, K.S.; Flavell, R.A.; Marks, C.B.; Miyake, K.; Liston, D.R.; Fowler, K.T.; Gorelick, F.S.; Andrews, N.W. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J. Cell Biol. 2003, 162, 543–549. [Google Scholar] [CrossRef]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [Green Version]
- LaPlante, J.M.; Sun, M.; Falardeau, J.; Dai, D.; Brown, E.M.; Slaugenhaupt, S.A.; Vassilev, P.M. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol. Genet. Metab. 2006, 89, 339–348. [Google Scholar] [CrossRef]
- Paczkowski, J.E.; Richardson, B.C.; Fromme, J.C. Cargo adaptors: structures illuminate mechanisms regulating vesicle biogenesis. Trends Cell Biol. 2015, 25, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Renard, H.F.; Johannes, L.; Morsomme, P. Increasing Diversity of Biological Membrane Fission Mechanisms. Trends Cell Biol. 2018, 28, 274–286. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Autophagic lysosome reformation. Exp. Cell Res. 2013, 319, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Patel, B.; Aphkhazava, D.; Macian, F.; Santambrogio, L.; Shields, D.; Cuervo, A.M. The lipid kinase PI4KIIIbeta preserves lysosomal identity. EMBO J. 2013, 32, 324–339. [Google Scholar] [CrossRef]
- Bright, N.A.; Gratian, M.J.; Luzio, J.P. Endocytic delivery to lysosomes mediated by concurrent fusion and kissing events in living cells. Curr. Biol. CB 2005, 15, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Heuser, J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J. Cell Biol. 1989, 108, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Zaarur, N.; Meriin, A.B.; Bejarano, E.; Xu, X.; Gabai, V.L.; Cuervo, A.M.; Sherman, M.Y. Proteasome failure promotes positioning of lysosomes around the aggresome via local block of microtubule-dependent transport. Mol. Cell. Biol. 2014, 34, 1336–1348. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Neefjes, J. Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 2017, 47, 1–8. [Google Scholar] [CrossRef]
- Harada, A.; Takei, Y.; Kanai, Y.; Tanaka, Y.; Nonaka, S.; Hirokawa, N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998, 141, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.J. Emerging mechanisms of dynein transport in the cytoplasm versus the cilium. Biochem. Soc. Trans. 2018, 46, 967–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasa, M.A.; Maximiano, F.C.; Smolarczyk, K.; Francis, R.E.; Betson, M.E.; Lozano, E.; Goldenring, J.; Seabra, M.C.; Rak, A.; Ahmadian, M.R.; et al. Armus is a Rac1 effector that inactivates Rab7 and regulates E-cadherin degradation. Curr. Biol. CB 2010, 20, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa-Ferreira, C.; Munro, S. Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell 2011, 21, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Loubery, S.; Wilhelm, C.; Hurbain, I.; Neveu, S.; Louvard, D.; Coudrier, E. Different microtubule motors move early and late endocytic compartments. Traffic 2008, 9, 492–509. [Google Scholar] [CrossRef]
- Bentley, M.; Decker, H.; Luisi, J.; Banker, G. A novel assay reveals preferential binding between Rabs, kinesins, and specific endosomal subpopulations. J. Cell Biol. 2015, 208, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Schindler, C.; Jia, R.; Jarnik, M.; Backlund, P.; Bonifacino, J.S. BORC, a multisubunit complex that regulates lysosome positioning. Dev. Cell 2015, 33, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Willett, R.; Martina, J.A.; Zewe, J.P.; Wills, R.; Hammond, G.R.V.; Puertollano, R. TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat. Commun. 2017, 8, 1580. [Google Scholar] [CrossRef] [Green Version]
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat. Commun. 2019, 10, 5630. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Mochida, K. Reticulophagy and nucleophagy: New findings and unsolved issues. Autophagy 2015, 11, 2377–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, M.; Sakai, Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J. 2010, 277, 3289–3294. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Dall’Armi, C.; Devereaux, K.A.; Di Paolo, G. The role of lipids in the control of autophagy. Curr. Biol. CB 2013, 23, R33–R45. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef]
- Zamani, M.; Taher, J.; Adeli, K. Complex role of autophagy in regulation of hepatic lipid and lipoprotein metabolism. J. Biomed. Res. 2017, 31, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Drizyte, K.; Casey, C.A.; McNiven, M.A. Hepatic Lipophagy: New Insights into Autophagic Catabolism of Lipid Droplets in the Liver. Hepatol. Commun. 2017, 1, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Hammerling, B.C.; Gustafsson, A.B. Mitochondrial quality control in the myocardium: cooperation between protein degradation and mitophagy. J. Mol. Cell Cardiol. 2014, 75, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Lievens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [Green Version]
- Shires, S.E.; Gustafsson, A.B. Mitophagy and heart failure. J. Mol. Med. Berl. 2015, 93, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Q.; Li, Y.; Li, C.; Zhu, Y.; Xia, F.; Xu, S.; Li, W. PTEN-Induced Putative Kinase 1 (PINK1)/Parkin-Mediated Mitophagy Protects PC12 Cells Against Cisplatin-Induced Neurotoxicity. Med. Sci. Monit. 2019, 25, 8797–8806. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Peng, H.; Yang, J.; Li, G.; You, Q.; Han, W.; Li, T.; Gao, D.; Xie, X.; Lee, B.H.; Du, J.; et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017, 27, 657–674. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J. Mol. Biol. 2020, 432, 231–239. [Google Scholar] [CrossRef]
- Radulovic, M.; Schink, K.O.; Wenzel, E.M.; Nahse, V.; Bongiovanni, A.; Lafont, F.; Stenmark, H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, C.; Meyer, H. Detection and Clearance of Damaged Lysosomes by the Endo-Lysosomal Damage Response and Lysophagy. Curr. Biol. CB 2017, 27, R1330–R1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Salvati, A.; Boya, P. Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. Lausanne 2018, 9, 778. [Google Scholar] [CrossRef]
- Patel, B.; Cuervo, A.M. Methods to study chaperone-mediated autophagy. Methods 2015, 75, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Koller, A.; Rangell, L.K.; Keller, G.A.; Subramani, S. Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J. Cell Biol. 1998, 141, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Seo, A.Y.; Lau, P.W.; Feliciano, D.; Sengupta, P.; Gros, M.A.L.; Cinquin, B.; Larabell, C.A.; Lippincott-Schwartz, J. AMPK and vacuole-associated Atg14p orchestrate mu-lipophagy for energy production and long-term survival under glucose starvation. eLife 2017, 6. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Dersh, D.; Iwamoto, Y.; Argon, Y. Tay-Sachs disease mutations in HEXA target the alpha chain of hexosaminidase A to endoplasmic reticulum-associated degradation. Mol. Biol. Cell 2016, 27, 3813–3827. [Google Scholar] [CrossRef]
- Raben, N.; Plotz, P.; Byrne, B.J. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr. Mol. Med. 2002, 2, 145–166. [Google Scholar] [CrossRef]
- Mellies, U.; Lofaso, F. Pompe disease: A neuromuscular disease with respiratory muscle involvement. Respir. Med. 2009, 103, 477–484. [Google Scholar] [CrossRef] [Green Version]
- McCall, A.L.; Salemi, J.; Bhanap, P.; Strickland, L.M.; Elmallah, M.K. The impact of Pompe disease on smooth muscle: a review. J. Smooth Muscle Res. 2018, 54, 100–118. [Google Scholar] [CrossRef] [Green Version]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Platt, F.M. Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic 2010, 11, 419–428. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol. Dis. 2016, 90, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Evans, E.; Pelled, D.; Riebeling, C.; Bodennec, J.; de-Morgan, A.; Waller, H.; Schiffmann, R.; Futerman, A.H. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 2003, 278, 23594–23599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkotian, E.; Schwarz, A.; Pelled, D.; Schwarzmann, G.; Segal, M.; Futerman, A.H. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 1999, 274, 21673–21678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelled, D.; Lloyd-Evans, E.; Riebeling, C.; Jeyakumar, M.; Platt, F.M.; Futerman, A.H. Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J. Biol. Chem. 2003, 278, 29496–29501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Appelqvist, H.; Sandin, L.; Bjornstrom, K.; Saftig, P.; Garner, B.; Ollinger, K.; Kagedal, K. Sensitivity to lysosome-dependent cell death is directly regulated by lysosomal cholesterol content. PLoS ONE 2012, 7, e50262. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 1888–1896. [Google Scholar] [CrossRef]
- Ahmed, I.; Liang, Y.; Schools, S.; Dawson, V.L.; Dawson, T.M.; Savitt, J.M. Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 16503–16509. [Google Scholar] [CrossRef]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain J. Neurol. 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Harding, R.J.; Tong, Y.F. Proteostasis in Huntington’s disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chow, C.Y.; Sahenk, Z.; Shy, M.E.; Meisler, M.H.; Li, J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain J. Neurol. 2008, 131, 1990–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Martyn, C.; Li, J. Fig4 deficiency: A newly emerged lysosomal storage disorder? Prog. Neurobiol. 2013, 101–102, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergin, I.; Evans, T.D.; Razani, B. Degradation and beyond: the macrophage lysosome as a nexus for nutrient sensing and processing in atherosclerosis. Curr. Opin. Lipidol. 2015, 26, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef] [Green Version]
- Javaheri, A.; Bajpai, G.; Picataggi, A.; Mani, S.; Foroughi, L.; Evie, H.; Kovacs, A.; Weinheimer, C.J.; Hyrc, K.; Xiao, Q.; et al. TFEB activation in macrophages attenuates postmyocardial infarction ventricular dysfunction independently of ATG5-mediated autophagy. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.C.; Bartlett, J.J.; Perez, L.J.; Brunt, K.R.; Legare, J.F.; Hassan, A.; Kienesberger, P.C.; Pulinilkunnil, T. Glucolipotoxicity diminishes cardiomyocyte TFEB and inhibits lysosomal autophagy during obesity and diabetes. Biochim. Biophys. Acta 2016, 1861, 1893–1910. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Stypmann, J.; Janssen, P.M.; Prestle, J.; Engelen, M.A.; Kogler, H.; Lullmann-Rauch, R.; Eckardt, L.; von Figura, K.; Landgrebe, J.; Mleczko, A.; et al. LAMP-2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic Res. Cardiol. 2006, 101, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, S.; Shiomi, K.; Yamamoto, A.; Nishino, I.; Nonaka, I.; Ohi, T. LAMP-2 positive vacuolar myopathy with dilated cardiomyopathy. Intern. Med. 2007, 46, 757–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. https://doi.org/10.3390/cells9051131
Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells. 2020; 9(5):1131. https://doi.org/10.3390/cells9051131
Chicago/Turabian StyleTrivedi, Purvi C., Jordan J. Bartlett, and Thomas Pulinilkunnil. 2020. "Lysosomal Biology and Function: Modern View of Cellular Debris Bin" Cells 9, no. 5: 1131. https://doi.org/10.3390/cells9051131
APA StyleTrivedi, P. C., Bartlett, J. J., & Pulinilkunnil, T. (2020). Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells, 9(5), 1131. https://doi.org/10.3390/cells9051131