Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice

 ,
,  ,
,  ,
, 
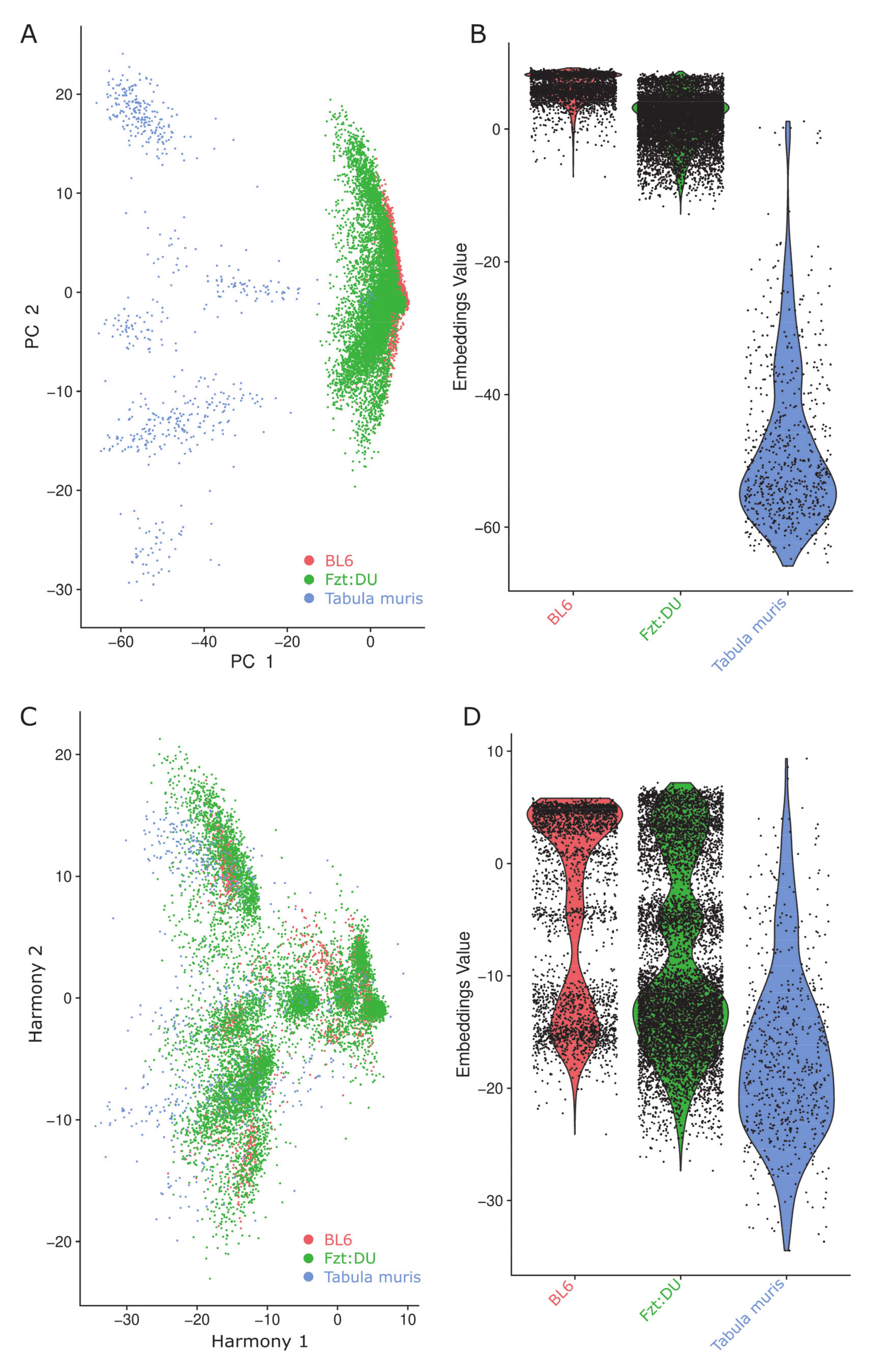{kind=link}
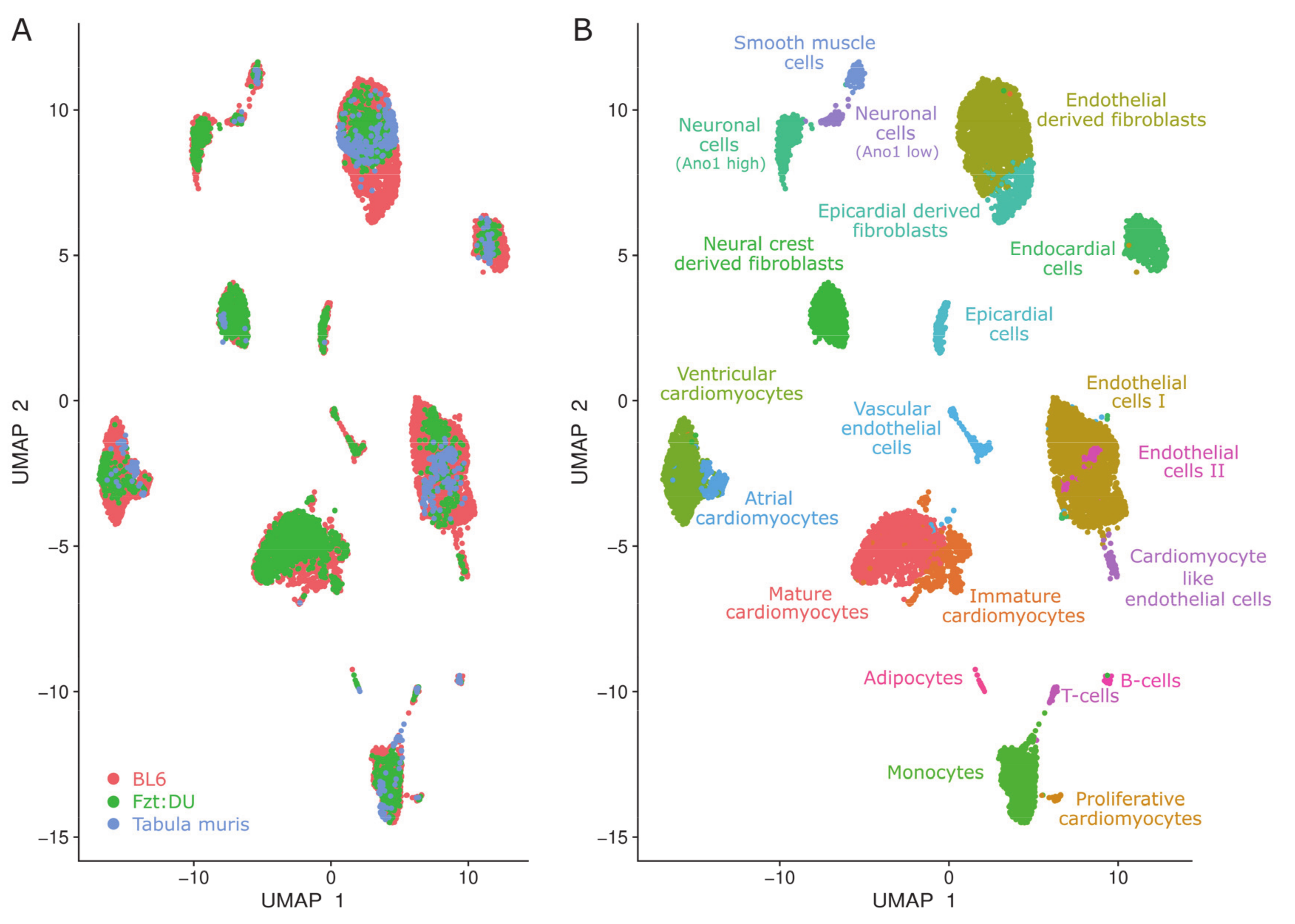{kind=link}
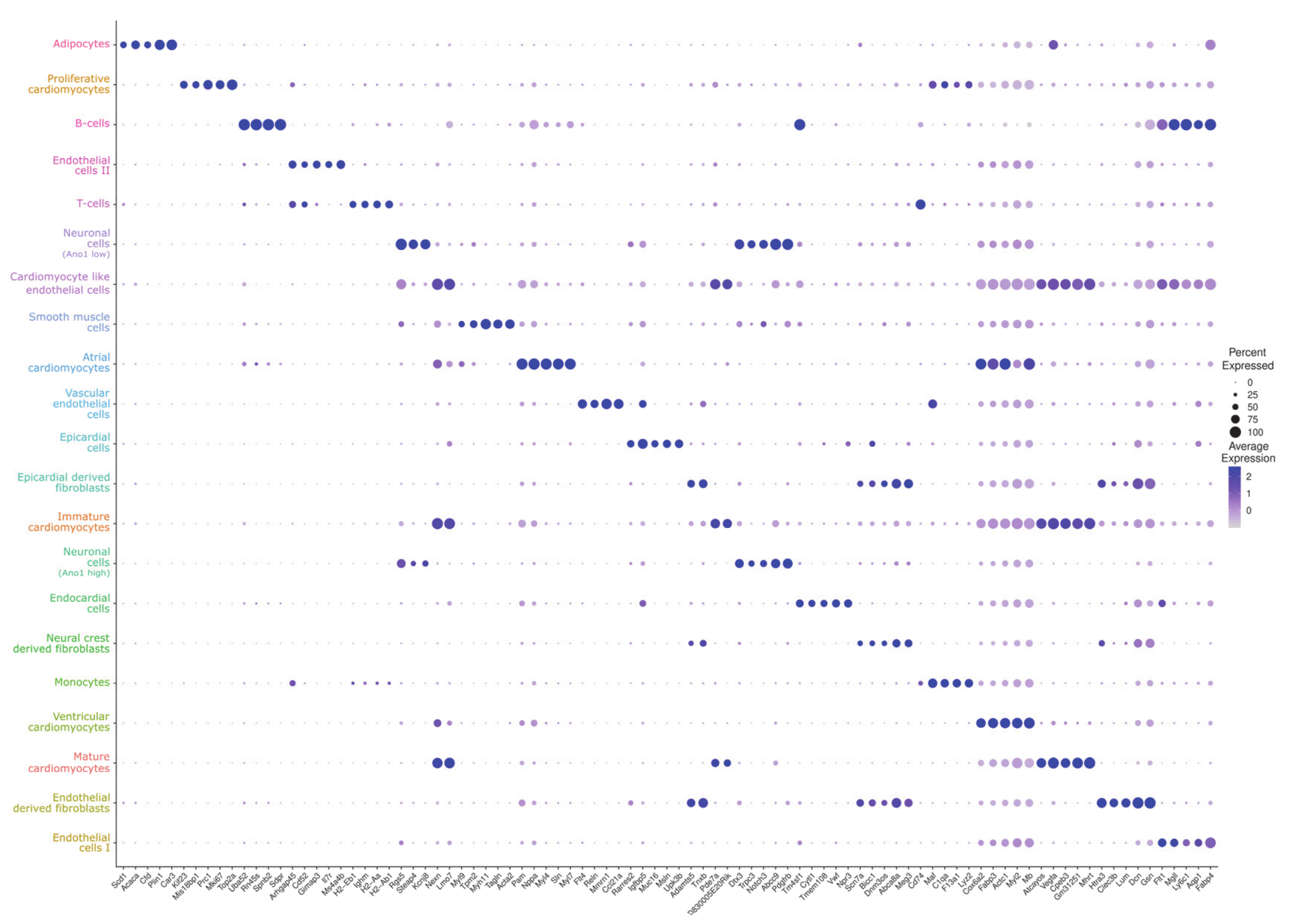{kind=link}
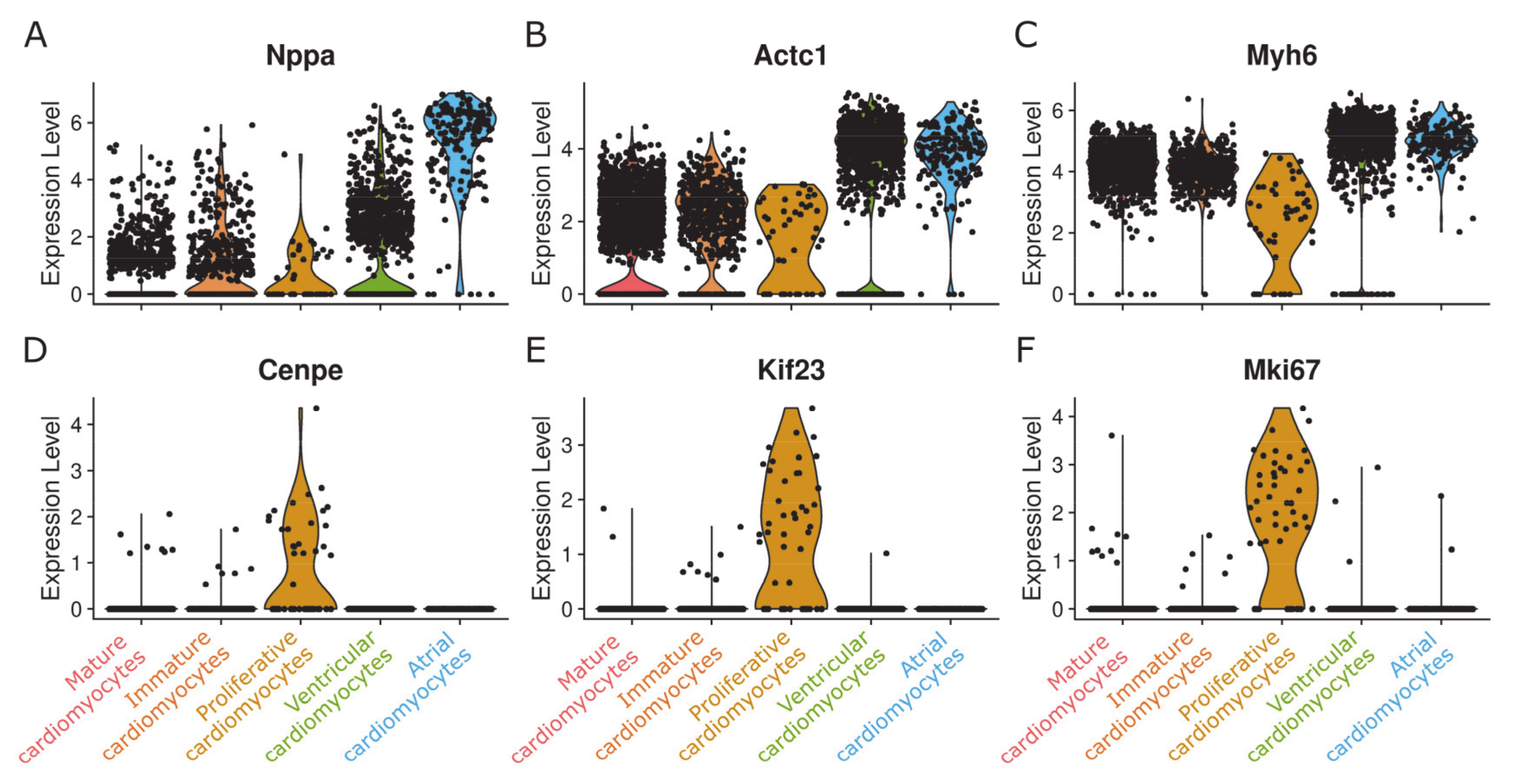{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation of Single Cells and Single Nuclei
2.3. Single-Cell and Single-Nucleus Sequencing
2.4. Computational Data Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, P.; Liu, J.; Zhao, J.; Wilkins, B.J.; Lupino, K.; Wu, H.; Pei, L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular Regulation of Cardiomyocyte Differentiation. Circ. Res. 2015, 116, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Mäkinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484. [Google Scholar] [CrossRef]
- Yavari, A.; Bellahcene, M.; Bucchi, A.; Sirenko, S.; Pinter, K.; Herring, N.; Jung, J.J.; Tarasov, K.V.; Sharpe, E.J.; Wolfien, M.; et al. Mammalian γ2 AMPK regulates intrinsic heart rate. Nat. Commun. 2017, 8, 1–19. [Google Scholar] [CrossRef]
- Gladka, M.M.; Molenaar, B.; de Ruiter, H.; van der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.A.; Huibers, M.M.H.; van Oudenaarden, A.; van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [Google Scholar] [CrossRef]
- Massaia, A.; Chaves, P.; Samari, S.; Miragaia, R.J.; Meyer, K.; Teichmann, S.A.; Noseda, M. Single Cell Gene Expression to Understand the Dynamic Architecture of the Heart. Front. Cardiovasc. Med. 2018, 5, 167. [Google Scholar] [CrossRef] [Green Version]
- Picelli, S. Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol. 2017, 14, 637. [Google Scholar] [CrossRef] [Green Version]
- Wirka, R.C.; Pjanic, M.; Quertermous, T. Advances in Transcriptomics. Circ. Res. 2018, 122, 1200–1220. [Google Scholar] [CrossRef]
- Wolfien, M.; Galow, M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Goldammer, T.; Wolkenhauer, O.; Hoeflich, A.; David, R. Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells 2020, 9, E318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkinopoulos, I.; Ishida, H.; Saba, R.; Ruchaya, P.; Cabrera, C.; Struebig, M.; Barnes, M.; Terry, A.; Kaneko, M.; Shintani, Y.; et al. Single-Cell Expression Profiling Reveals a Dynamic State of Cardiac Precursor Cells in the Early Mouse Embryo. PLoS ONE 2015, 10, e0140831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.K.; Kent, D.G.; Buettner, F.; Shehata, M.; Macaulay, I.C.; Calero-Nieto, F.J.; Sánchez Castillo, M.; Oedekoven, C.A.; Diamanti, E.; Schulte, R.; et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 2015, 16, 712. [Google Scholar] [CrossRef] [Green Version]
- Kernfeld, E.M.; Genga, R.M.; Neherin, K.; Magaletta, M.E.; Xu, P.; Maehr, R. A single-cell transcriptomic atlas of thymus organogenesis resolves cell types and developmental maturation. Immunity 2018, 48, 1258. [Google Scholar] [CrossRef] [Green Version]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- The Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372.
- Ackers-Johnson, M.; Li, P.Y.; Holmes, A.P.; O’Brien, S.-M.; Pavlovic, D.; Foo, R.S. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ. Res. 2016, 119, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Ackers-Johnson, M.; Tan, W.L.W.; Foo, R.S.-Y. Following hearts, one cell at a time: Recent applications of single-cell RNA sequencing to the understanding of heart disease. Nat. Commun. 2018, 9, 4434. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Lake, B.B.; Codeluppi, S.; Yung, Y.C.; Gao, D.; Chun, J.; Kharchenko, P.V.; Linnarsson, S.; Zhang, K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017, 7, 6031. [Google Scholar] [CrossRef] [PubMed]
- Linscheid, N.; Logantha, S.J.R.J.; Poulsen, P.C.; Zhang, S.; Schrölkamp, M.; Egerod, K.L.; Thompson, J.J.; Kitmitto, A.; Galli, G.; Humphries, M.J.; et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat. Commun. 2019, 10, 2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selewa, A.; Dohn, R.; Eckart, H.; Lozano, S.; Xie, B.; Gauchat, E.; Elorbany, R.; Rhodes, K.; Burnett, J.; Gilad, Y.; et al. Systematic Comparison of High-throughput Single-Cell and Single-Nucleus Transcriptomes during Cardiomyocyte Differentiation. Sci. Rep. 2020, 10, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Kirita, Y.; Donnelly, E.L.; Humphreys, B.D. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 23–32. [Google Scholar] [CrossRef]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Jiang, S.; Kong, X.; El-Ali, N.; Ball, A.R.; Ma, C.I.-H.; Hashimoto, N.; Yokomori, K.; Mortazavi, A. Single-nucleus RNA-seq of differentiating human myoblasts reveals the extent of fate heterogeneity. Nucleic Acids Res. 2016, 44, e158. [Google Scholar] [CrossRef] [Green Version]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef]
- González-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E.; Burns, C.G. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 2018, 44, 433.e7–446.e7. [Google Scholar] [CrossRef] [Green Version]
- Bersell, K.; Arab, S.; Haring, B.; Kühn, B. Neuregulin1/ErbB4 Signaling Induces Cardiomyocyte Proliferation and Repair of Heart Injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, A.H.; Philip, V.M.; Chesler, E.J.; Mogil, J.S. Comparing phenotypic variation between inbred and outbred mice. Nat. Methods 2018, 15, 994–996. [Google Scholar] [CrossRef]
- Wanner, M. The Unexpected Advantages of Outbred Mice in Research. Available online: https://www.jax.org/news-and-insights/2018/december/the-advantages-of-outbred-mice-in-research (accessed on 6 February 2020).
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11.14.1–11.14.19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888.e21–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Wolfien, M.; Galow, M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Goldammer, T.; Wolkenhauer, O.; Hoeflich, A.; David, R. Single Nuclei Sequencing of entire Mammalian Hearts: Strain-dependent Cell Type Composition and Velocity. Cardiovasc. Res. 2020, cvaa054. [Google Scholar] [CrossRef]
- Atilla-Gokcumen, G.E.; Castoreno, A.B.; Sasse, S.; Eggert, U.S. Making the cut: The chemical biology of cytokinesis. ACS Chem. Biol. 2010, 5, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Normand, G.; King, R.W. Understanding Cytokinesis Failure. Adv. Exp. Med. Biol. 2010, 676, 27–55. [Google Scholar]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997, 272, H220–H226. [Google Scholar] [CrossRef]
- Bakken, T.E.; Hodge, R.D.; Miller, J.A.; Yao, Z.; Nguyen, T.N.; Aevermann, B.; Barkan, E.; Bertagnolli, D.; Casper, T.; Dee, N.; et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 2018, 13, e0209648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levkau, B.; Schäfers, M.; Wohlschlaeger, J.; von Wnuck Lipinski, K.; Keul, P.; Hermann, S.; Kawaguchi, N.; Kirchhof, P.; Fabritz, L.; Stypmann, J.; et al. Survivin Determines Cardiac Function by Controlling Total Cardiomyocyte Number. Circulation 2008, 117, 1583–1593. [Google Scholar] [CrossRef] [Green Version]
- Sheng, L.; Wan, B.; Feng, P.; Sun, J.; Rigo, F.; Bennett, C.F.; Akerman, M.; Krainer, A.R.; Hua, Y. Downregulation of Survivin contributes to cell-cycle arrest during postnatal cardiac development in a severe spinal muscular atrophy mouse model. Hum. Mol. Genet. 2018, 27, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Yester, J.W.; Kühn, B. Mechanisms of Cardiomyocyte Proliferation and Differentiation in Development and Regeneration. Curr. Cardiol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázár, E.; Sadek, H.A.; Bergmann, O. Cardiomyocyte renewal in the human heart: Insights from the fall-out. Eur. Heart J. 2017, 38, 2333–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zeng, C.; et al. Dedifferentiation, Proliferation, and Redifferentiation of Adult Mammalian Cardiomyocytes After Ischemic Injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparative analysis of single cell RNA-sequencing methods. bioRxiv 2019, 632216. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galow, A.-M.; Wolfien, M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Hoeflich, A.; Wolkenhauer, O.; David, R.; Goldammer, T. Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice. Cells 2020, 9, 1144. https://doi.org/10.3390/cells9051144
Galow A-M, Wolfien M, Müller P, Bartsch M, Brunner RM, Hoeflich A, Wolkenhauer O, David R, Goldammer T. Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice. Cells. 2020; 9(5):1144. https://doi.org/10.3390/cells9051144
Chicago/Turabian StyleGalow, Anne-Marie, Markus Wolfien, Paula Müller, Madeleine Bartsch, Ronald M. Brunner, Andreas Hoeflich, Olaf Wolkenhauer, Robert David, and Tom Goldammer. 2020. "Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice" Cells 9, no. 5: 1144. https://doi.org/10.3390/cells9051144
APA StyleGalow, A. -M., Wolfien, M., Müller, P., Bartsch, M., Brunner, R. M., Hoeflich, A., Wolkenhauer, O., David, R., & Goldammer, T. (2020). Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice. Cells, 9(5), 1144. https://doi.org/10.3390/cells9051144