As detailed in
Supplemental Table S1, evidence for nuclear receptor interaction was found in the literature for 20 agriculturally used azoles: bitertanol, bromuconazole, cyproconazole, difeconazole, etaconazole, epoxiconazole, fenbuconazole, flusilazole, hexaconazole, imazalil, myclobutanil, prochloraz, propiconazole, prothioconazole, tebuconazole, thiabendazole, triadimefon, triadimenol, triflumizole, and uniconazole. Gained depth of information was highly variable: for a few compounds, sparse literature data were available only from a single publication addressing one single endpoint, e.g., AHR interaction by bitertanol, difeconazole, prothioconazole, and triadimenol [
25,
26,
27], whereas other compounds were covered by several publications together addressing all three receptors of interest. Most extensive research with respect to nuclear receptor activation has been performed with cyproconazole, epoxiconazole, imazalil, prochloraz, propiconazole, and tebuconazole (
Supplemental Table S1). The latter compounds were therefore selected for in-depth discussion below. Accordingly, the following tables accompanying the main text of this work contain the details for these compounds. Effects are sorted by species (mouse, rat, human), system used for investigation (cell-free, in vitro, in vivo), and effect type (activation/induction indicated by ↑; inhibition/repression indicated by ↓; reported no-effect findings not mentioned here but in
Supplemental Table S1). Further details are presented in
Supplemental Table S1.
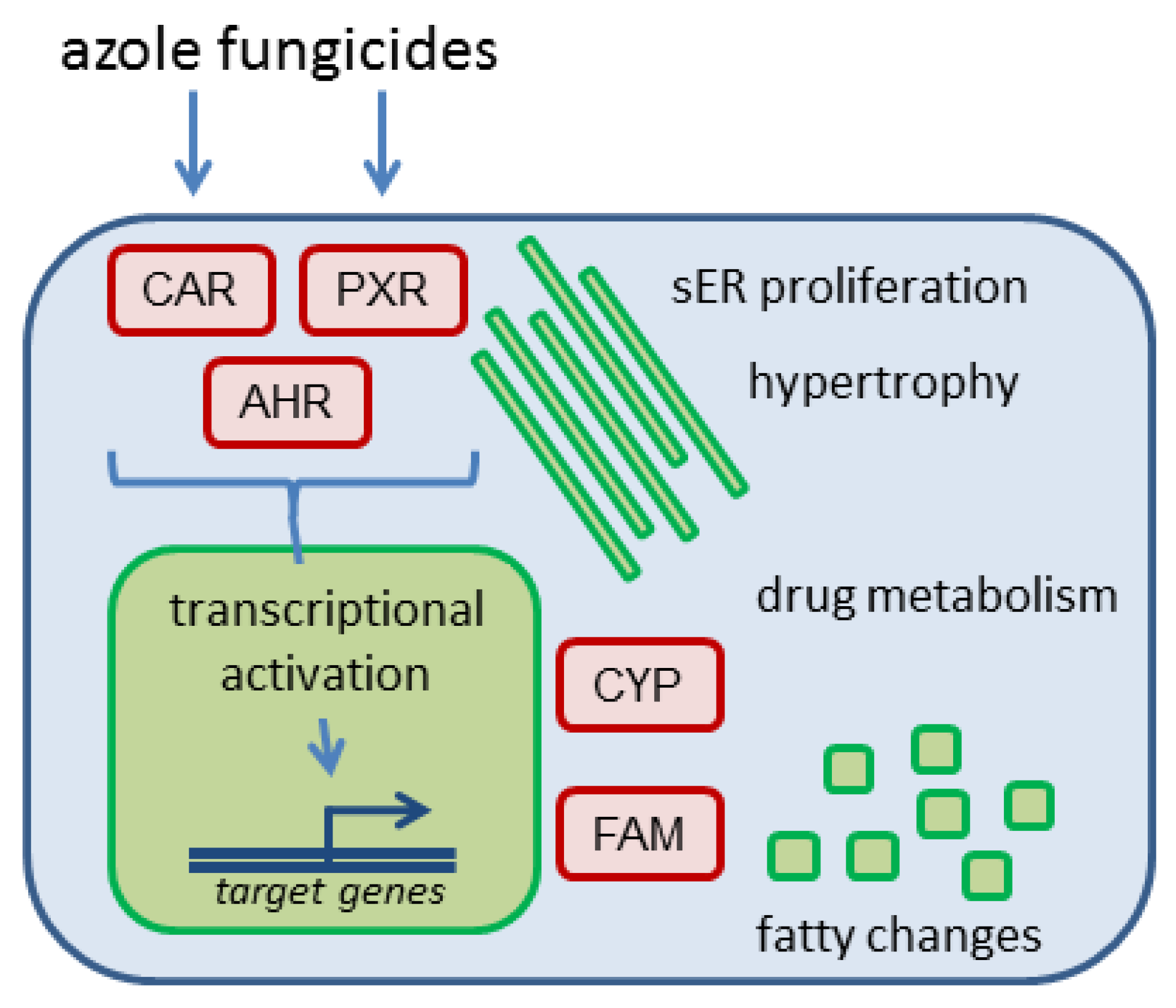
In general, mostly indirect evidence for nuclear receptor interaction (i.e., in the form of target gene or protein expression) comes from in vivo studies. Mice and rats were the species of choice in most studies (
Supplemental Table S1), whereas individual papers also describe azole effects in fish species [
27,
28,
29]. In some studies, more direct evidence for involvement of a certain receptor is presented due to the use of knockout or humanized mouse strains [
9,
10,
18,
19,
20,
30]. Human hepatocellular cell systems have frequently been used for in vitro studies, while also data from rat or mouse primary hepatocytes or permanent liver cell lines constitute an important part of the available in vitro information. In addition, non-liver human cell lines such as Caco-2, Jeg-3, HeLa, and HEK293 have been employed in some studies, while monkey COS-1 or COS-7 cells were selected in some studies to analyze the nuclear receptors from other species in reporter gene assays (
Supplemental Table S1). The bandwidth of in vitro endpoints ranges from FRET assays of nuclear receptor binding to various luciferase reporter gene systems and, similar to the in vivo studies, target gene and protein expression. Especially by using specific constructs in reporter gene assays, in vitro data contribute the bulk of direct evidence for interaction of a certain azole fungicide with one of the nuclear receptors of interest. Computational approaches such as molecular docking or toxicogenomic pathway analyses have played a role in a minority of past projects to identify the nuclear receptor-interacting potential of azoles [
6,
31,
32,
33]. As a rule, the vast majority of effects observed were receptor activation and subsequent target gene induction. A lack of effects has also been reported sometimes, and these no-effect data are included in
Supplemental Table S1 as well. Inhibition of one of the nuclear receptors AHR, CAR and PXR by an azole fungicide appears to be a rather rare event and has been reported only for tebuconazole and bromuconazole with CAR [
16,
32,
34], as well as for bitertanol and prothioconazole with AHR [
25]. Additional inhibiting effects occur with respect to the activities of target enzymes from the CYP family [
32,
35]. These observations, however, are most likely caused by direct inhibition of mammalian CYP enzymes rather than to nuclear receptor-mediated effects, given the fact the respective enzymes were demonstrated to be upregulated at the transcriptional and/or protein levels and in line with the original design of azoles as fungal CYP51 inhibitors. In the following, the compounds for which most data are available are discussed in detail and hepatic effects observed in regulatory in vivo studies are related to the known nuclear receptor-activating properties of the compounds.
3.1. Propiconazole
Available literature suggests that propiconazole is a weak activator of mouse, rat, and human AHR (
Table 1). Data for mouse and rat are mainly derived from in vivo studies that yielded indirect evidence for AHR activation via target gene and enzyme activity regulation [
4,
6,
31,
36]. For human liver cells, in vitro evidence at the reporter gene, target mRNA, protein, and enzyme activity level suggests weak agonism of propiconazole at the AHR, substantiated by in silico molecular docking study results [
16,
26,
27,
31,
35,
37,
38]. Additionally, in vitro experiments using a human AHR antagonist, AHR binding site-mutated reporter variants or AHR knockout cells mechanistically strengthen the evidence for AHR activation by propiconazole [
31].
Similarly, in vivo rat and mouse data at the target mRNA and enzyme activity levels indirectly connect propiconazole with the activation of CAR [
5,
6,
20,
33,
36,
39,
40] (
Table 2). This conclusion is supported also by in vitro reporter gene data including CAR binding site-deficient mutant constructs [
32,
33], and by the fact that CAR target gene induction was abolished in a CAR-deficient knockout mouse model [
20]. Propiconazole agonism at CAR appears not to be limited to murine systems, as in vitro FRET, reporter gene and target mRNA data as well as molecular docking show that CAR and its target genes are activated by the compound also in human cells [
16,
31,
32,
33,
38]. Activation of CAR appears to occur with rather moderate potency, as suggested by in vitro assays and computationally derived binding energy [
32]. Target enzyme activity inhibition in human cells in vitro is most likely due to an inhibition of the metabolic enzyme and not by a nuclear receptor-mediated mechanism [
32].
For PXR activation by propiconazole, in vivo rat and mouse data are only available at the target mRNA level (
Table 3), demonstrating upregulation of PXR target genes in both species [
4,
5,
6,
33,
36]. Specificity of this type of information is, of course, questionable because the target gene batteries of CAR and PXR show substantial overlap. In human cells, however, direct evidence at the reporter gene and mRNA level is available supporting the notion that propiconazole is a moderately potent agonist of human PXR [
12,
16,
28,
31,
32,
38]. This is substantiated by experiments in a CAR-deficient, PXR-expressing cell line [
32]. Again, target enzyme activity inhibition is presumably a consequence of direct CYP inhibition [
32,
35].
Propiconazole has been assessed for its toxicity after short- and long-term exposure as well as for its carcinogenic potential in vivo in a number of studies conducted according to harmonized OECD test guidelines within the regulatory approval procedure. Study results are summarized in the assessment report [
41], JMPR [
42], as well as in the respective EFSA conclusion [
43]. Within the renewal procedure of propiconazole it was decided not to approve this active substance to be used as a pesticide in the EU anymore also because it is classified as toxic to reproduction category 1B [
41]. Here effects observed on the target organ liver are summarized: the substance caused an increase in absolute and relative liver weights in short- and long-term rodent studies. The lowest NOAEL for hepatotoxicity was 3.6 mg/kg body weight per day in the chronic rat study. With respect to liver toxicity the following histopathological findings were observed: hepatocellular hypertrophy (rats and mice in short- and long-term studies), fatty changes (rats and mice in short- and long-term studies), hepatocellular cell degeneration (rats and mice in short- and long-term studies), and neoplasms (hepatocellular adenoma and carcinoma in mice). In addition, alterations in clinical chemistry were observed, supporting the abovementioned histopathological findings, namely increased ALT activity (mice) and altered γGT activity (rats, both after short- and long-term treatment).
The observations made in regulatory studies are in line with published scientific literature also demonstrating increased liver weight and hypertrophy in rats and mice [
4,
6,
20,
36], as well as altered expression of fatty acid metabolism-related genes also in rats and mice [
5,
6,
40]. Increases in liver weight and hepatocellular proliferation induced by propiconazole were abolished in
Car knockout mice demonstrating the important role of the receptor in the development of hepatotoxicity after propiconazole exposure [
20].
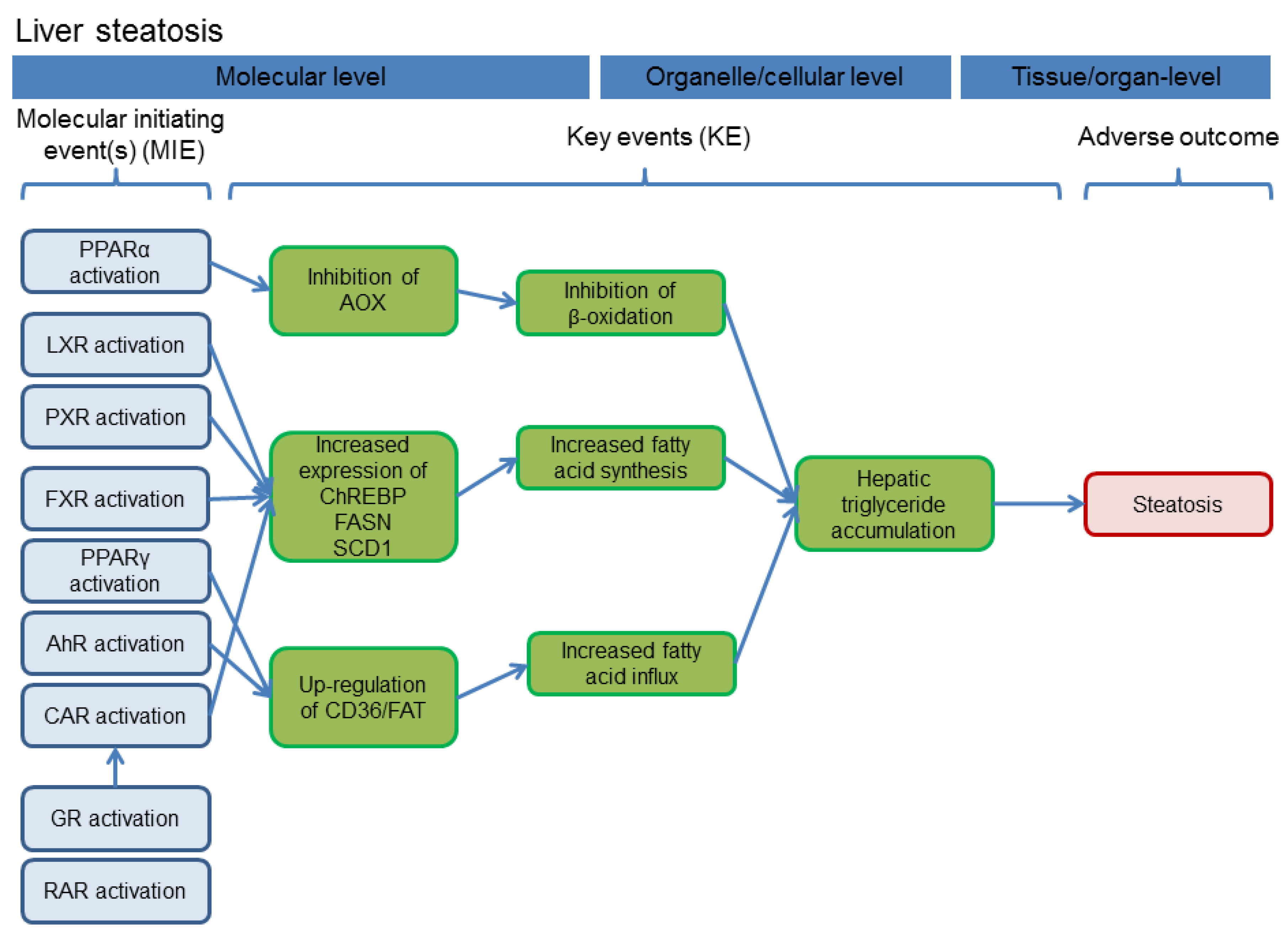
Nuclear receptor activation occurs as molecular initiating events of the pathway(s) leading to different adverse effects in the liver. For two important hepatic outcomes frequently observed after exposure to azole fungicides, i.e., hepatocellular hypertrophy and fatty changes/steatosis, schematic drawings of adverse outcome pathways (AOPs) are presented in
Figure 2 and
Figure 3, respectively.
Hepatocellular hypertrophy after xenobiotic exposure is [
44], similar to the induction of CYPs and other drug-metabolizing enzymes [
45], often observed in perivenous hepatocytes following activation of CAR and/or PXR (
Figure 2); for specific observations with different azoles please refer to the text below. This is plausible as perivenous hepatocytes possess higher levels of CAR and AHR, as compared to periportal hepatocytes [
46,
47], and also stronger endogenous activation of the canonical Wnt/β-catenin pathway which intensifies signal transduction via different xeno-sensing receptors [
48,
49,
50,
51,
52,
53,
54,
55,
56]. In hepatic steatosis, various nuclear receptors, including CAR, PXR, and AHR, play a major role in the etiology of the adverse outcome (
Figure 3; see also e.g., [
57,
58]). Additionally, long-term exposure to activators of CAR and AHR is known to lead to the formation of neoplasms as observed in the long-term rodent studies, as for example reviewed in [
59,
60]. Even though activation of AHR, CAR or PXR does not directly lead to hepatocellular cell degeneration, prolonged exposure to substances increasing the activity of enzymes such as the CYPs, known to produce reactive oxygen species and therefore to increase cellular stress, may contribute to cell degeneration.
Figure 3.
Schematic delineation of the AOP for hepatocellular steatosis. The figure was adapted from [
58]. Abbreviations: FXR, farnesoid-X-receptor, GR, glucocorticoid receptor.
Figure 3.
Schematic delineation of the AOP for hepatocellular steatosis. The figure was adapted from [
58]. Abbreviations: FXR, farnesoid-X-receptor, GR, glucocorticoid receptor.
Thus, several aspects of the adverse hepatic effects of propiconazole observed in vivo can be explained by the activation of the nuclear receptors CAR and PXR: this comprises the findings of hepatocellular hypertrophy resulting in an elevated liver weight (cp.
Figure 2), as well as the changes related to fatty acid metabolism (cp.
Figure 3). The study with
Car knockout mice underlines the role of this receptor in short-term effects of propiconazole exposure [
20], and it appears likely that also the formation of hepatic neoplasms as observed in long-term rodent studies links to tumor promotion following persistent activation of CAR. PXR activation might probably also be involved in the regulation of hepatocyte proliferation, but is currently not considered a relevant factor in liver tumor promotion [
61]. Comparison of the data available from human and rodent systems suggests that human hepatocytes react with similar receptor activation as mouse or rat hepatocytes. With respect to the downstream consequences, target gene activation related to xenobiotic metabolism is well documented in human cells (
Table 1,
Table 2 and
Table 3), and also the PXR-dependency of triglyceride accumulation in human HepaRG hepatoma cells upon propiconazole exposure is well-documented [
16]. Human relevance of long-term tumorigenic effects of the compound is more difficult to judge; it is not clear whether CAR activation is the sole driver of propiconazole-dependent tumorigenesis. Activation of CAR is often considered not to be relevant for human tumorigenesis [
59], even though this topic is still disputed [
62]. Nonetheless, without clear evidence that other tumorigenic mechanisms do not play a relevant role here, it remains challenging to draw a final conclusion. It should be noted, however, that non-genotoxic mechanisms of tumor induction are expected to be linked to the long-term presence of the tumor-promoting compound above a certain threshold level, and that exposure to minor residues of a compound, as for example consumer exposure via foodstuff, cannot be expected to necessarily fulfil these criteria.
3.2. Epoxiconazole
Limited evidence is available for activation of the AHR by epoxiconazole (
Table 4): weak activation of the receptor in different human cell lines is suggested by findings at the target mRNA and protein levels [
26,
63,
64], whereas no substantial induction of a human AHR-driven reporter assays system has been observed in a human placental cell line [
63]. Gene expression and enzyme activity data from in vivo studies and from cultured cells also suggest weak AHR activation by epoxiconazole in the rat [
64,
65,
66].
Data on the effects of epoxiconazole on CAR are mostly based only on indirect findings at the target mRNA and enzyme activity levels obtained from in vivo studies (
Table 5). In mice, induction of
Cyp2b10 transcription, and also the analysis of a broader CAR-dependent gene expression signature indicate activation of the receptor by epoxiconazole [
20,
39]. Similarly, the induction of CAR target CYPs in the livers of epoxiconazole-treated rats suggests CAR activation in that species [
65,
66]. The latter studies provide additional evidence for CAR activation in rat liver by showing increased activity of CAR target CYPs [
65,
66]. Induction of a human CAR-dependent reporter assay system has been reported in one publication [
64].
A comparable picture emerges when it comes to PXR activation by epoxiconazole (
Table 6): again, only indirect data from CYP induction is available from in vivo studies. Here, PXR target mRNA expression and enzyme activity were increased in rats, suggesting PXR agonism of the compound [
65,
66]. No data have been published for other species.
Epoxiconazole has been assessed for its toxicity after short- and long-term exposure as well as for its carcinogenic potential in vivo in a number of studies conducted according to harmonized OECD test guidelines within the regulatory approval procedure. Study results are summarized in the respective EFSA conclusion [
67] and in the assessment report on the active substance epoxiconazole [
68]. Epoxiconazole is approved as a pesticide active substance in the EU. However, due to classification as developmental and reproductive toxicant category 1B the renewal is pending. In this review, only effects observed on the target organ liver are considered. The substance caused increase in absolute and relative liver weights in short- and long-term rodent studies. The lowest NOAEL for hepatotoxicity was 0.8 mg/kg body weight per day in the chronic rat study. With respect to liver toxicity, the following histopathological findings were observed in the regulatory studies: hepatocellular hypertrophy (mice and rats in short- and long-term studies), fatty changes (mice and rats in short- and long-term studies), hepatocellular degeneration (mice and rats in short- and long-term studies), liver inflammation (dogs, short-term treatment) and neoplasms (hepatocellular adenomas and carcinomas in mice after long-term treatment). In addition, alterations in clinical chemistry were observed, supporting the abovementioned histopathological findings, namely increased activity of γGT and other parameters in rodents after short- and long-term treatment.
The findings are in line with observations published in the scientific literature also describing increased liver weight and hypertrophy upon epoxiconazole exposure in rat and mouse liver [
39,
65,
66,
69]. In mice, also proliferation of hepatocytes has been reported [
39]. Analyses of mRNA expression have demonstrated altered expression of fatty acid metabolism-related genes in epoxiconazole-treated rat livers [
66].
Hepatotoxic effects observed after exposure to epoxiconazole in vivo can be explained by the activation of nuclear receptors as molecular initiating events of toxicity. Even if the majority of data consists of indirect evidence from target gene or protein induction studies, it is possible to conclude that epoxiconazole activates CAR and PXR and thus facilitates the development of hepatocellular hypertrophy (cp.
Figure 2) as well as of fatty changes/steatosis (cp.
Figure 3). Nuclear receptor activation is often an underlying cause also for hepatocellular tumorigenesis, and the available data indicating that epoxiconazole is only a very weak activator of AHR make it appear likely that a CAR-mediated carcinogenic mechanism may be more relevant here. Possible human relevance of CAR-mediated liver tumor formation has been addressed above. Nonetheless, the available data cannot unequivocally rule out contributions by other nuclear receptors or via additional pathways not involving nuclear receptors. The activation of AHR, CAR, or PXR does not directly lead to hepatocellular cell degeneration. Prolonged exposure to substances increasing activity of enzymes such as the CYPs, however, produces reactive oxygen species and therefore increase cellular stress. This in turn may contribute to hepatocellular degeneration. Inflammatory effects, by contrast, are most likely not due to the activation of the receptors alone. Instead, these findings may base upon processes occurring secondary to cellular damage.
3.3. Cyproconazole
Evidence for activation of the AHR by cyproconazole appears inconsistent (
Table 7). Elevated AHR target gene expression was observed in mouse liver, whereas results at the protein level did not show consistent upregulation [
9,
10,
30]. In addition, in rat liver and in a rat liver cell line increased AHR target mRNA expression was observed, but this effect was not reflected in changes at the target enzyme activity level [
64,
65,
66]. Three studies in human cells showed no induction of AHR-dependent reporter gene systems [
17,
30,
63], while data on target gene induction are inconsistent between cell lines showing no or moderate induction [
17,
63,
64]. In summary, cyproconazole is no or only a very weak activator of AHR-dependent signal transduction.
As with AHR, cyproconazole appears not to activate human CAR to a major extent: even though upregulation of target gene mRNA expression has been reported in human liver cells (
Table 8), reporter gene assays did not reveal substantial activation of CAR by cyproconazole in different studies [
17,
30,
64]. Target gene expression only should not be regarded as strong evidence due to the overlapping target gene batteries of CAR and PXR. By contrast, in vivo studies have shown that cyproconazole is an activator of mouse CAR, as evidenced by target mRNA, protein, and enzyme activity [
9,
10,
19,
20,
30,
39]. Importantly, experiments with CAR-deficient mice have shown that CAR is substantially involved in the observed hepatic effects [
9,
10,
19]. In addition, the fact that humanized CAR/PXR mice showed smaller target gene and enzyme induction than wildtype mice further strengthens the conclusion that cyproconazole activity at CAR shows considerable species differences [
30]. In rats, the compound also seems to activate CAR, as suggested by elevated target mRNA and enzyme activity levels [
65,
66].
PXR activation in human liver cells has been clearly shown at the reporter gene, as well as at the target mRNA and protein levels [
17]. No in vitro and/or mechanistic data are available to clarify the possible PXR activation by cyproconazole in rodents, but elevated target mRNA expression in mice and rats [
9,
10,
65,
66], as well as increased target enzyme activity in both species [
19,
65,
66] suggest that PXR activation by cyproconazole is not limited to human cells (
Table 9).
Cyproconazole has been assessed for its toxicity after short- and long-term exposure as well as for its carcinogenic potential in vivo in a number of studies conducted according to harmonized OECD test guidelines within the regulatory approval procedures. Study results are summarized in the respective EFSA conclusion [
70] and in the Assessment Report on the active substance cyproconazole [
71]. Cyproconazole is approved as a pesticide active substance in the EU. In the following, effects observed on the target organ liver are presented: the substance caused an increase in absolute and relative liver weights in short- and long-term rodent studies as well as in short-term dog studies [
71]. The lowest NOAEL for hepatotoxicity was 2 mg/kg body weight per day in the chronic rat study. With respect to liver toxicity the following histopathological findings were observed: hepatocellular hypertrophy (rats, mice, dogs), fatty changes (rats, mice), hepatocellular cell degeneration (rats, mice), liver inflammation, as well as neoplasms (hepatocellular adenomas and carcinomas in mice after long-term treatment). In addition, alterations in clinical chemistry were observed, supporting the abovementioned histopathological findings, namely increased ALAT (rats, dogs) and γGT activities (rats).
Published data from non-regulatory studies support the above findings: increased liver weight and hypertrophy have been reported for cyproconazole-treated rats and mice [
9,
10,
19,
30,
39,
65,
66,
69]. Hepatotoxicity in mice is also underlined by increased ALAT levels [
19], and proliferative responses have been documented in mouse liver [
9,
19,
30]. Vacuolization indicative of fatty acid changes was observed in rats and mice [
9,
19,
30,
65,
66]. At the mRNA level, cyproconazole-induced alterations in genes related to fatty acid metabolism and transport have been demonstrated in rats and mice [
30,
66]. A partial role of CAR in liver effects caused by cyproconazole has been substantiated by analyses in CAR-deficient mice showing diminished responses with respect to liver weight, hypertrophy, proliferation, fat vacuolization and liver tumor development in the knockout strain, as compared to wildtype mice [
9,
10,
19]. Cyproconazole exerted reduced but still detectable effects on the abovementioned endpoints in
Car knockout mice, and thus it has been concluded that both, CAR-dependent as well as CAR-independent mechanisms are responsible for liver hypertrophy and liver tumor development in cyproconazole-treated mice [
10]. Species differences in hepatotoxicity are suggested by a study with humanized CAR/PXR mice showing that hepatocellular proliferation and vacuolization was absent in the humanized animals, whereas an increase in liver weight was still observed [
30].
The activation of nuclear receptors as molecular initiating events may explain most of the hepatotoxic effects of cyproconazole observed in vivo. Mechanistic analyses with
Car knockout mice have demonstrated the importance of the receptor in vivo for liver hypertrophy as well as for fatty acid changes [
9,
10,
19] (cp.
Figure 2 and
Figure 3). Effects observed in human cells appear to be mostly mediated by PXR, as the compound is obviously no potent activator of human CAR. Interestingly, the abovementioned studies have concluded that cyproconazole appears to exert a minor fraction of its effects related to liver hypertrophy, fatty acid changes, proliferation, and tumorigenesis via CAR-independent mechanisms. Thus, even in the case that (i) CAR-dependent tumorigenesis is considered not relevant for humans and that (ii) CAR is not substantially activated in human cells by cyproconazole, some uncertainty remains with respect to the possibility of the possible human relevance of other tumorigenic mechanisms. It should be noted that some of the hepatotoxic effects of cyproconazole were also observed in dogs, making the underlying mechanism less likely to be rodent-specific. The scenario that multiple pathways contribute to liver toxicity (as for example CAR-dependent and independent mechanisms for cyproconazole) may also apply to other (tri)azole fungicides. Unfortunately, only very few compounds have been studied using nuclear receptor-knockout or -transgenic animals to provide a convincing experimental basis for that assumption. As noted above, nuclear receptor activation does not directly lead to hepatocellular cell degeneration, but reactive oxygen species generated during prolonged exposure to CYP inducers may cause such degenerative effects. Inflammatory effects, on the other hand, may not be directly explained by activation of the receptors alone and rather constitutes a secondary finding.
3.4. Tebuconazole
AHR activity of tebuconazole is an interesting case: on the one hand, reporter gene assays in human cell lines failed to reveal increased AHR activity [
26,
37,
63]. On the other hand, analyses at the target mRNA, protein, and enzyme activity levels showed a clear increase [
26,
35,
37,
38,
63]. This induction was abolished in AHR-KO cells or after pharmacological inhibition of AHR, demonstrating that the process of target gene induction is AHR-dependent [
37]. Future research will help to clarify the exact molecular mechanisms by which tebuconazole affects AHR-dependent transcription. Only few data have been published on AHR activation by tebuconazole in other species (
Table 10). Indirect evidence from target mRNA and enzyme activity studies suggests AHR activation in mouse and rat liver [
10,
37].
Unexpected findings have also been reported with respect to the influence of tebuconazole on CAR (
Table 11): molecular docking and FRET analyses suggest that the compound is binding to human CAR [
32], and interestingly reporter gene assays have shown that tebuconazole is a potent CAR inhibitor in vitro [
16,
32]. Irrespective of the inhibition of CAR in human cells, CAR target mRNA expression and enzyme activity were shown to be increased by tebuconazole in different studies [
32,
37,
38]. This obvious discrepancy may be explained by the fact that CAR and PXR targets show considerable overlap and thus the observed induction is most likely a consequence of simultaneous PXR activation in the cells (see also below). The observed increase in CAR-dependent reporter gene signals in rat primary hepatocytes, by contrast, suggested an agonistic potential at rat CAR that was verified using a mutant CAR binding site-deficient reporter variant [
32]. Further verification was provided by demonstration of tebuconazole-increased target mRNA expression in rat hepatocytes [
32]. In mice, CAR target mRNA and protein expression data suggest that mouse CAR is also activated by tebuconazole [
9,
10].
With respect to PXR activation, indirect evidence from target gene expression analysis in mice points towards an agonistic behavior of tebuconazole at the receptor [
10], while no data are available for the rat (
Table 12). In human cells, in vitro analyses with different reporter systems have demonstrated that tebuconazole activates PXR, and experimental prove has been provided showing that this effect is independent from CAR [
16,
32]. Activation of PXR is further supported by findings at the mRNA and enzyme activity levels showing increased transcript levels or substrate conversion upon tebuconazole treatment of human liver cells [
32,
37,
38]. However, it should be noted that no change in PXR-dependent CYP enzyme activity has been detected in a human intestinal cell line [
35].
Tebuconazole has been assessed for its toxicity after short- and long-term exposure as well as for its carcinogenic potential in vivo in a number of studies conducted according to harmonized OECD test guidelines within the regulatory approval procedure. Study results are summarized in the respective JMPR evaluation [
72] and EFSA conclusion [
73]. Tebuconazole is approved as a pesticide active substance in the EU. Here effects observed on the target organ liver are presented. The substance caused increases in absolute and relative liver weights in short- and long-term rodent studies. The lowest NOAEL for hepatotoxicity was 16 mg/kg body weight per day in the chronic rat study. With respect to liver toxicity the following histopathological findings were observed: hepatocellular hypertrophy (rats and mice in short- and long-term studies), fatty changes (rats and mice in short- and long-term studies), hepatocellular cell degeneration (rats and mice in short- and long-term studies), liver inflammation (rats and mice in short- and long-term studies), neoplasms (adenomas and carcinomas in mice after long-term treatment) and lesions of the biliary epithelium (rats and mice in short- and long-term studies). In addition, alterations in clinical chemistry were observed, supporting the abovementioned histopathological findings, namely an increase in activity of AST, ALT and γGT in some dog short-term and rodent short- and long-term studies.
In line with the findings from regulatory studies, tebuconazole has caused increased liver weight, hepatocellular hypertrophy, vacuolization and proliferation in mice [
9,
10]. Moreover, the compound caused tumor development in mouse liver [
10]. When comparing wildtype with
Car knockout mice, it became obvious that tumor induction was more or less abolished in the knockout strain [
10], whereas the genotype differences were not that pronounced for hepatocellular hypertrophy [
9,
10]. From these data, it was concluded that CAR plays a crucial role for tumor development by tebuconazole in mouse liver, whereas other receptors, potentially including PXR, are mostly responsible for the hypertrophic response [
10].
As discussed above for epoxiconazole, propiconazole and cyproconazole, findings on liver hypertrophy (cp.
Figure 2) and fatty acid changes (cp.
Figure 3) following exposure to tebuconazole may be well explained by the nuclear receptor activation profile of the compound, demonstrating CAR and PXR induction in rodent hepatocytes. Similarly, as also discussed above, cell degeneration may relate to long-term nuclear receptor activation and subsequent CYP-dependent generation of reactive oxygen species, whereas inflammatory processes should probably regarded as not directly nuclear receptor-mediated. While the tumorigenic mechanism in mice is CAR-dependent, other effects such as hypertrophy appear to relate to PXR activation [
10]. This connects to the situation in human cells where changes in triglyceride accumulation occur in a PXR-dependent manner, as evidenced by mechanistic investigations [
16]. CAR-mediated tumor induction by tebuconazole is most probably not to be considered relevant for humans, because the compound has been demonstrated a CAR antagonist in human cells [
16,
32].
3.5. Prochloraz
For prochloraz, activation of AHR has been shown in different species (
Table 13). In cultured human cells, the compound activated AHR-driven luciferase reporter systems [
30,
63] and also AHR target mRNA expression [
26,
63]. AHR target gene induction has been also observed in rat liver, along with elevated target enzyme activity [
65,
66]. In mice, prochloraz is able to increase hepatic AHR target mRNA and protein expression [
30] The extent of that effect was, with respect to Cyp1a1 induction, similar between wildtype and CAR/PXR-humanized mice, indicating that prochloraz is a comparable agonist of mouse and human AHR [
30]. At the protein level, total CYP1A content increased more in wildtype mice, suggesting a possible contribution of murine CAR via the induction of
Cyp1a2 [
30].
CAR target mRNA and protein expression in mouse liver following exposure to prochloraz was also analyzed in the aforementioned study (
Table 14). Here, the inducing effects of prochloraz were much more pronounced in wildtype mice, as compared to their CAR/PXR-humanized counterparts [
30]. This indicates that prochloraz is more potent at mouse CAR, as compared to the human receptor. Nonetheless, in vitro stimulation of a reporter assay in HC-AFW1 cells in vitro by prochloraz also suggests some activity in human cells [
30]. In rats, data obtained at the CAR target mRNA and enzyme activity levels suggest activation of the receptor by prochloraz [
65,
66].
The limited amount of available data for prochloraz and PXR (
Table 15) shows only weak inducing effects in rat liver with respect to target mRNA and enzyme activity [
65,
66]. In human cells, activation of a PXR-dependent reporter assay system has been shown [
12].
Prochloraz has been assessed for its toxicity after short- and long-term exposure as well as for its carcinogenic potential in vivo in a number of studies conducted according to harmonized OECD test guidelines within the regulatory approval procedure. Study results are summarized in the respective EFSA conclusion [
74]. Prochloraz is approved as a pesticide active substance in the EU. Here effects observed on the target organ liver are presented. The substance caused increase in absolute and relative liver weights in short- and long-term rodent studies. The lowest NOAEL for hepatotoxicity was 1.3 mg/kg body weight per day in the chronic rat study. With respect to liver toxicity the following histopathological findings were observed: hepatocellular hypertrophy (rats, mice and dogs in short- and rats and mice in long-term studies), fatty changes (rats and mice in long-term studies), hepatocellular cell degeneration (rats and mice in short- and long-term studies), and the development of neoplasms (hepatocellular adenomas and carcinomas in mice in long-term studies),
The above findings are substantiated by results from published scientific studies demonstrating elevated liver weight and a hypertrophic response in prochloraz-treated rat and mouse livers [
30,
65,
66,
69]. Alterations in the expression of fatty acid metabolism-related genes have been observed in rat livers [
66].
Prochloraz is clearly an activator of the AHR in human and rodent cells, which makes the compound different from the above (tri)azoles which, if at all, activate the AHR only to a very limited degree. In addition, CAR and PXR also appear to be activated to a certain degree. In the absence of mechanistic studies, it is not possible to conclude the individual contributions of the different receptors to the development of hepatotoxicity. Hypertrophic responses (cp.
Figure 2), fatty acid changes (cp.
Figure 3) and cell degeneration may be related to CAR and PXR activation, but also to induction of AHR-dependent transcription. It should be noted that some of the effects were also observed in dogs, making the mechanisms less likely to be rodent-specific. In addition, with respect to tumorigenicity, the role of the individual receptors has not been elucidated which makes it challenging to conclude on possible human relevance of the findings. CAR activation in rodents is regularly followed by a pronounced transient proliferative response (e.g., see [
54,
75]). The role of AHR in tumor promotion seems to consist mainly in the inhibition of apoptosis, rather than in inducing proliferation [
76,
77,
78]. According to a poster abstract from a conference in 2015, prochloraz induces proliferation in wildtype mice, but not in mice with
Car knockout or expressing the human receptor (available at
www.toxicology.org/pubs/docs/Tox/2015Tox.pdf; p.351 of the document). This was interpreted by the authors as proof for a CAR-mediated, not human-relevant mechanism of tumorigenicity. Such a conclusion, however, appears premature: even independent of the discussion of human relevance of CAR-mediated tumor promotion, the mere observation of CAR activation and subsequent transient proliferation in short-term experiments has, if at all, only very limited predictive value with respect to the carcinogenic outcome. If a test compound simultaneously activates another, CAR-independent mechanism of tumor promotion that is linked to suppression of apoptosis in pre-malignant lesions rather than to transient proliferation in normal hepatocytes (see references above).
3.6. Imazalil
Imazalil should be regarded as a moderate activator of AHR in human liver cells (
Table 16). The compound induced AHR targets at the mRNA as well as at the protein level in vitro [
26,
38,
58], even though reporter assays did not reveal a substantial potential in human hepatoma cells [
58]. In a human intestinal cell line, target enzyme activity was induced, whereas only a weak tendency for increased
CYP1A1 transcription was visible [
35]. Induction of the AHR target
Cyp1a2 mRNA was observed in mice; however, the fact that the effect was diminished in Car-KO mice indicates a substantial contribution of the latter receptor to this finding [
18].
CAR activation by imazalil (
Table 17) has been extensively studied in human liver cells revealing that the receptor is activated by imazalil at the reporter gene assay [
58], target mRNA [
38,
58], and target protein levels [
58]. These findings are corroborated by results from mouse in vivo studies demonstrating elevated CAR target mRNA as well as protein levels in mouse liver following exposure to imazalil [
18,
79].
Reporter gene assays in human cell lines have demonstrated induction of PXR by imazalil [
12,
58] (
Table 18). Reporter gene analyses in human cells transfected with mouse PXR show activation also of mouse PXR [
79]. Accordingly, PXR target mRNA as well as protein expression is elevated by imazalil in human liver cells [
38,
58]. PXR target mRNA expression has also been analyzed in livers from imazalil-treated mice showing the expected induction [
18,
79]. Of note, inhibition of PXR target enzyme activity by imazalil has been reported in human intestinal cells [
35]. This observation, however, is likely a consequence of direct enzyme inhibition rather than of inhibitory effects on PXR.
Imazalil has been assessed for its toxicity after short- and long-term exposure as well as for its carcinogenic potential in vivo in a number of studies conducted according to harmonized OECD test guidelines within the regulatory approval procedure. Study results are summarized in the respective JMPR evaluation [
80] and EFSA conclusion [
81]. Imazalil is approved as a pesticide active substance in the EU. Here effects observed on the target organ liver are presented. The substance caused increase in absolute and relative liver weights in short- and long-term rodent studies. The lowest NOAEL for hepatotoxicity was 2.5 mg/kg body weight per day in the 1-year dog study supported by the 2-year rat study. With respect to liver toxicity the following histopathological findings were observed: hepatocellular hypertrophy (rats, mice and dogs in short- and long-term studies), fatty changes (rats and mice in short- and long-term studies), hepatocellular cell degeneration (rats and mice in short- and long-term studies), liver inflammation and neoplasms (hepatocellular adenomas and carcinomas in mice, and adenomas in rats after long-term treatment). In addition, alterations in clinical chemistry were observed, supporting the abovementioned histopathological findings, namely alterations in the activities of AST and ALT, and an increase in the activity of γGT in some rodent short- and long-term studies.
Hepatotoxicity of imazalil has also been addressed in other than regulatory studies: a very recent publication reported increase liver weight and hepatocellular hypertrophy in rats [
82]. In mice, increased liver weight has been observed in two studies [
18,
79]. Liver weight increase and hypertrophy after imazalil exposure were similar between wildtype and Car knockout mice, whereas liver tumor development was substantially diminished in the knockout group [
18]. From these data it was concluded that CAR drives tumor development after imazalil treatment, whereas most likely PXR plays a major role in the hypertrophic response [
18]. Of note, hepatocellular proliferation in mice following administration of a CAR agonist is boosted by simultaneous treatment with imazalil, indicating a cross-talk between CAR and PXR [
79].
Hepatic effects observed after administration of imazalil on the target organ liver in vivo can be explained by the activation of nuclear receptors as molecular initiating events, as for example pictured in
Figure 2 and
Figure 3. Imazalil is a multi-receptor agonist and the different receptors activated by the compound may all contribute to the observed findings. In addition, activation of the retinoic acid receptor (RAR) α and inhibition of peroxisome proliferator-activated receptor (PPAR) α may have contributed to fatty acid changes upon imazalil treatment [
58]. Nonetheless, it should be noted that the latter findings on receptor activation have been obtained with human cells and therefore the situation in rodent might differ. CAR activation appears to play an important role for liver tumor development, but not for the hypertrophic response [
18], which indicates similarities with the liver toxicity profiles of other azole compounds (see above).
3.7. Other Agricultural Azole Fungicides
Evidence for nuclear receptor activation is also available for a number of additional agricultural azole fungicides. Several compounds, however, have not been extensively studied and thus data are only available for rather few selected endpoints. Results for those compounds are briefly presented in
Table 19 and in the following text; for more details please refer to
Supplemental Table S1.
Bitertanol has shown its ability to inhibit an AHR-dependent reporter system in mice [
25]. Bromuconazole appears to be an inducer of PXR in rats in vivo, as evidenced by PXR target mRNA and protein induction, while effects of CAR appear to be minor or even inhibitory [
34]. Difeconazole induces the AHR target gene CYP1A1 in human hepatoma cells [
26]. In vitro, etaconazole has been screened for its activity at CAR from different species and the results suggest a weak activation of rat and canine CAR, whereas the human and mouse wildtype receptors were not substantially affected [
83]. Fenbuconazole is an in vitro activator of PXR in human cells [
12] and induces AHR-, CAR- and PXR-dependent CYP mRNAs in human primary hepatocytes [
38]. Flusilazole appears to be a moderate AHR activator in human cell lines, as suggested by reporter assay and target mRNA expression studies [
26,
63]. Hexaconazole is an inducer of AHR-, CAR-, and PXR-dependent target CYP mRNAs in human primary hepatocytes, as are triflumizole and uniconazole [
38]. Hypochondriazole exerts its activity even at sub-zero-molar concentrations [
84]. Little more data are available for myclobutanil: the compound activates AHR and according downstream target mRNA and protein expression in human cells [
5,
26,
27,
38] and is also an AHR inducer in medaka fish [
27]. Furthermore, mRNA expression data from rat and mouse liver as well as from cultured human hepatocytes suggest also the activation of CAR and PXR in these species [
5,
20,
38]. Prothioconazole has been shown to be an inhibitor of expression of the AHR target Cyp1a1 in a mouse hepatoma cell line [
25], while thiabendazole actived a mouse PXR-dependent reporter system in vitro [
79]. Available data for triadimefon point towards cross-species AHR activation by this compound in rats, mice and medaka fish in vivo [
5,
6,
27]. Human AHR appears to be also a target of triadimefon and additionally of triadimenol, as suggested by reporter assays with the human receptor in yeast cells [
27]. According to mRNA expression data obtained from mouse and rat livers, triadimefon is also capable of activating CAR and PXR in these species [
4,
5,
6,
20].





{kind=link}
{kind=link}
{kind=link}