Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain

 , and
, and 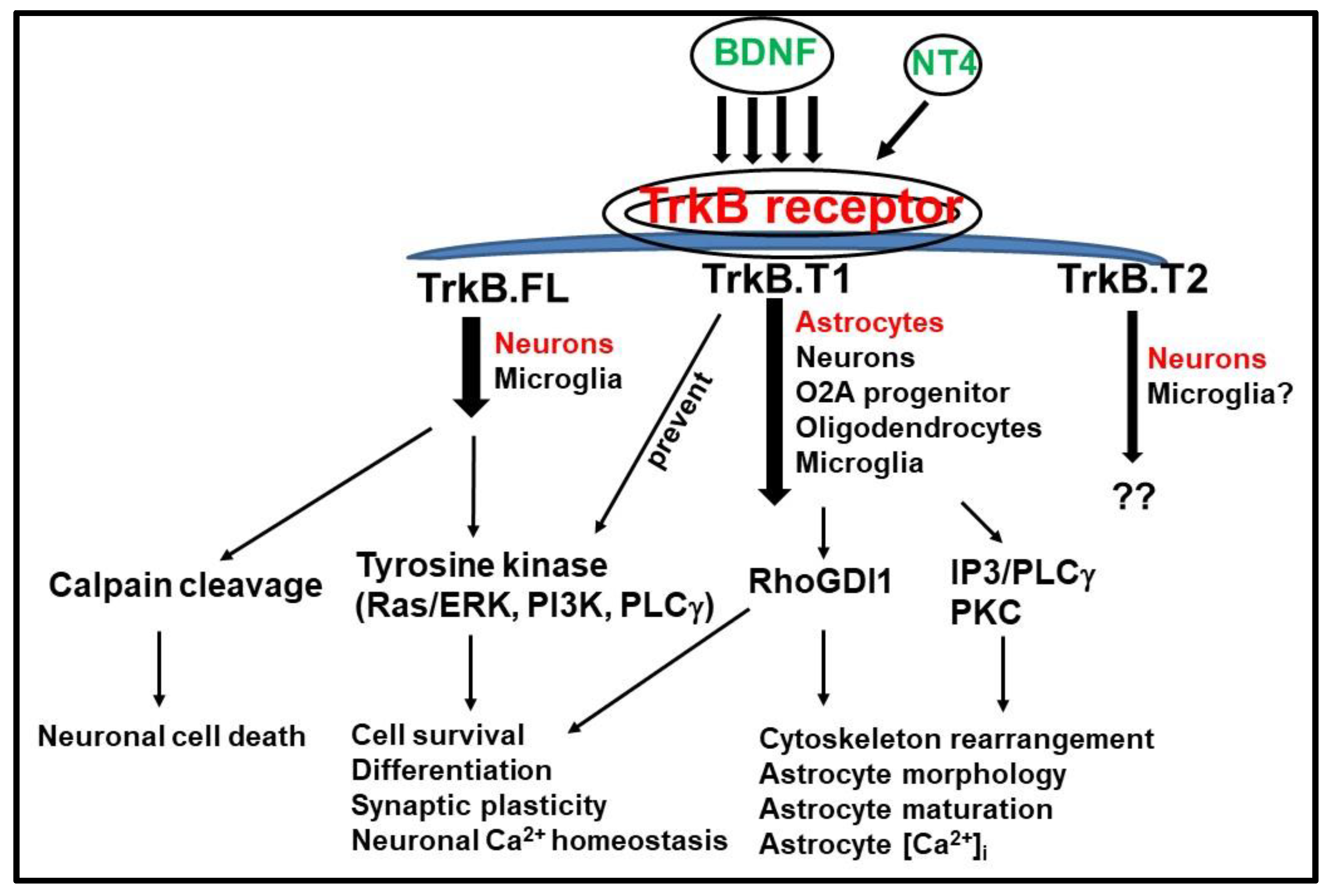{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. BDNF and TrkB Receptor Function during Development
3. TrkB.FL and TrkB.T1 Differential Signaling Pathways
3.1. TrkB.T1 Isoform as an Inhibitor of TrkB.FL
3.2. TrkB.T1 Signaling Pathway Independent of TrkB.FL
4. Function and Mechanisms of TrkB.T1 Receptor in CNS Insults
4.1. TrkB.T1 in Neurological Disorders
4.2. TrkB.T1 in Traumatic Spinal Cord Injury and Brain Injury
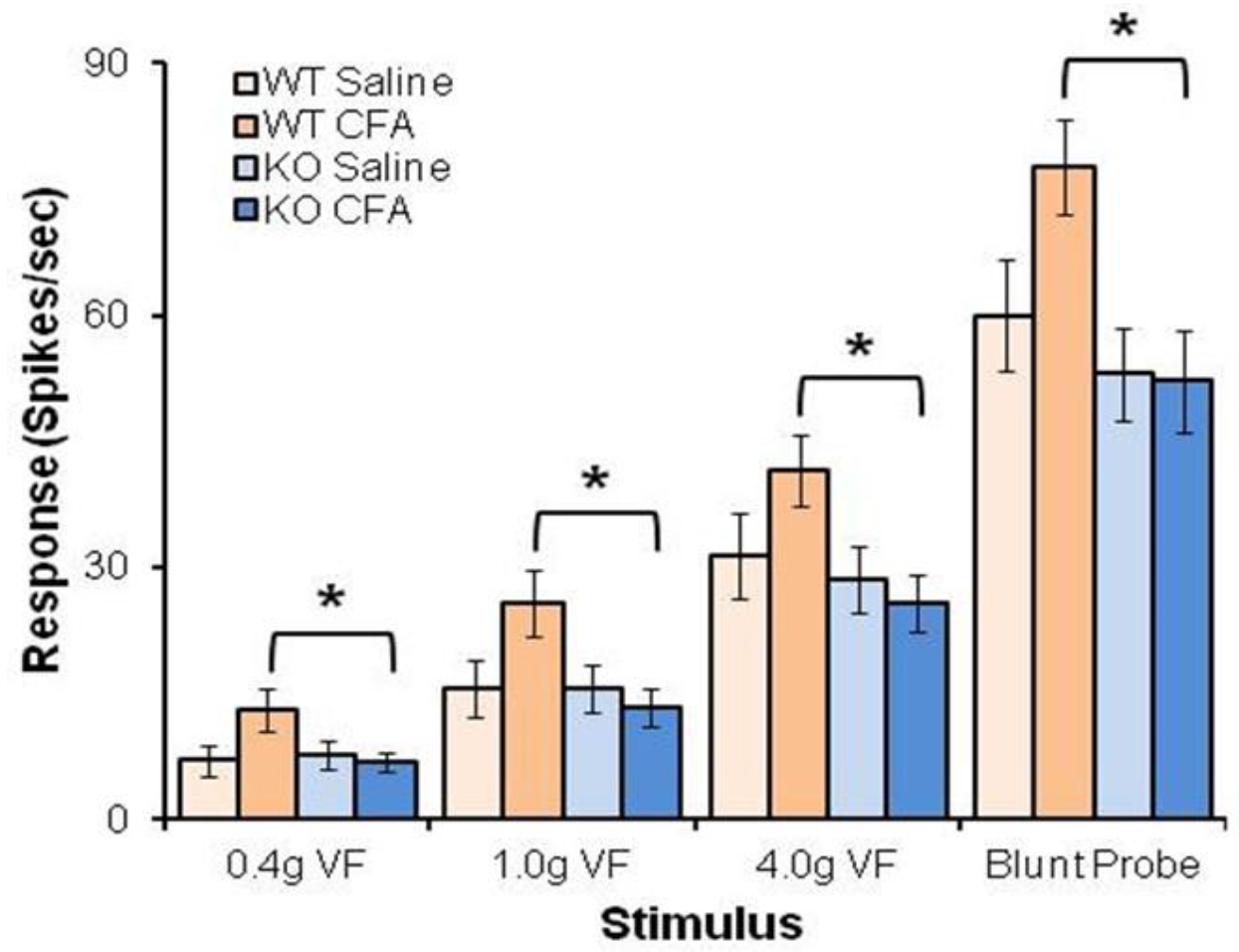
4.3. BDNF/TrkB.T1 Regulation in Neuropathic Pain
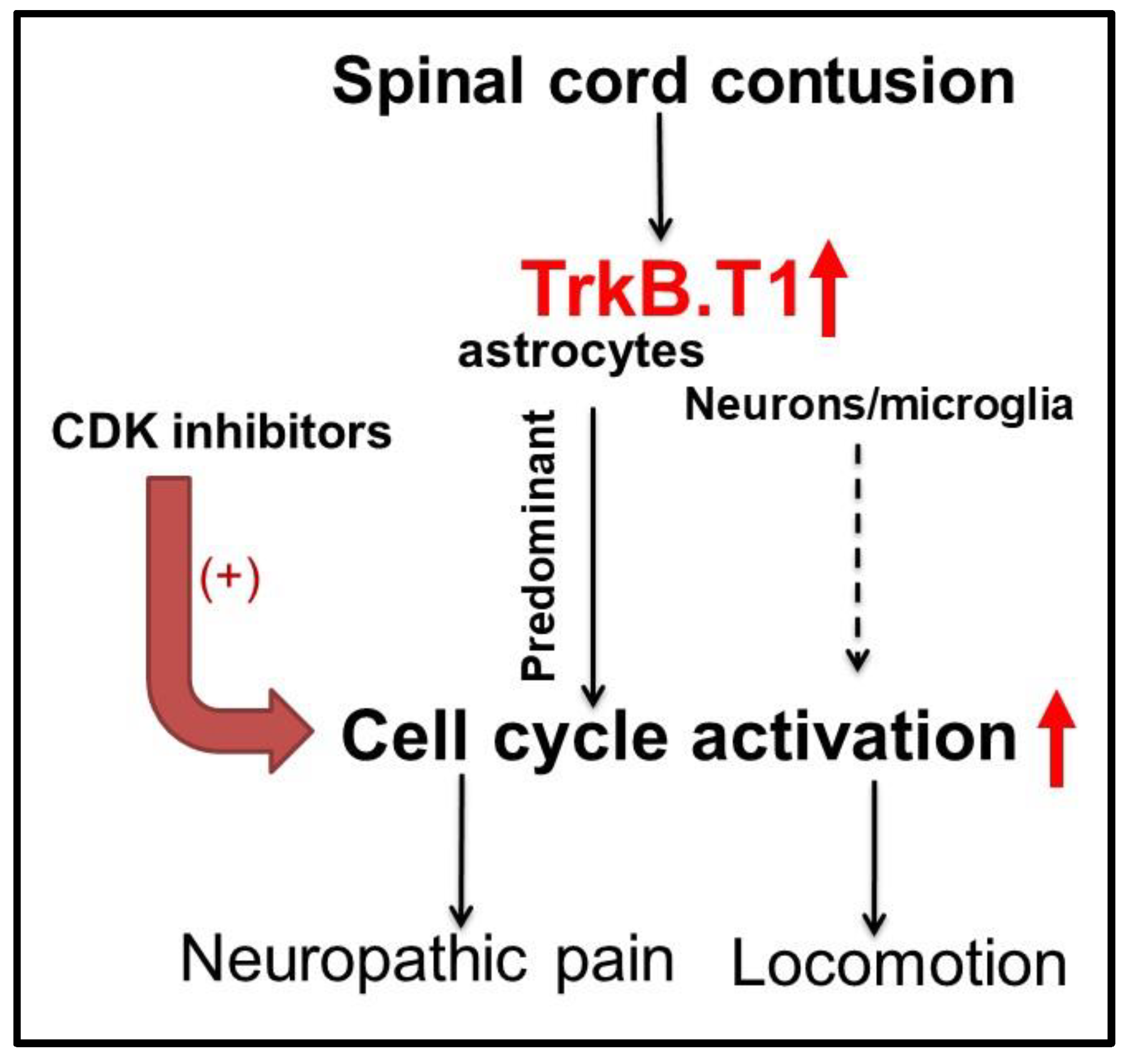
4.4. Astrocytic TrkB.T1 Contributes to Nociception via Dysregulated Cell Cycle Activation
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Yawn, B.P.; Wollan, P.C.; Weingarten, T.N.; Watson, J.C.; Hooten, W.M.; Melton, L.J., 3rd. The prevalence of neuropathic pain: Clinical evaluation compared with screening tools in a community population. Pain Med. 2009, 10, 586–593. [Google Scholar] [CrossRef] [Green Version]
- Vall, J.; Costa, C.M.; Santos Tde, J.; Costa, S.B. Neuropathic pain characteristics in patients from Curitiba (Brazil) with spinal cord injury. Arq. Neuropsiquiatr. 2011, 69, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Modirian, E.; Pirouzi, P.; Soroush, M.; Karbalaei-Esmaeili, S.; Shojaei, H.; Zamani, H. Chronic pain after spinal cord injury: Results of a long-term study. Pain Med. 2010, 11, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, D.D.; Bryce, T.N.; Shem, K.; Richards, J.S.; Elhefni, H. Gender and minority differences in the pain experience of people with spinal cord injury. Arch. Phys. Med. Rehab. 2004, 85, 1774–1781. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Robinson, J.E.; Ells, P.; Cole, J.D. Pain following spinal cord injury: Results from a postal survey. Pain 1988, 34, 101–102. [Google Scholar] [CrossRef]
- Siddall, P.J.; McClelland, J.M.; Rutkowski, S.B.; Cousins, M.J. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain 2003, 103, 249–257. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Johannesen, I.L.; Sindrup, S.H.; Bach, F.W.; Jensen, T.S. Pain and dysesthesia in patients with spinal cord injury: A postal survey. Spinal Cord 2001, 39, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Siddall, P.; Xu, C.L.; Cousins, M. Allodynia following traumatic spinal cord injury in the rat. Neuroreport 1995, 6, 1241–1244. [Google Scholar] [CrossRef]
- Siddall, P.J.; Cousins, M.J. Pain mechanisms and management: An update. Clin. Exp. Pharmacol. Physiol. 1995, 22, 679–688. [Google Scholar] [CrossRef]
- Siddall, P.J.; Taylor, D.; Cousins, M.J. Pain associated with spinal cord injury. Curr. Opin. Neurol. 1995, 8, 447–450. [Google Scholar] [CrossRef]
- Hulsebosch, C.E.; Hains, B.C.; Crown, E.D.; Carlton, S.M. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res. Rev. 2009, 60, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, R.; Keller, A. Chronic pain following spinal cord injury. Adv. Exp. Med. Biol. 2012, 760, 74–88. [Google Scholar] [PubMed] [Green Version]
- Thompson, S.W.; Bennett, D.L.; Kerr, B.J.; Bradbury, E.J.; McMahon, S.B. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc. Natl. Acad. Sci. USA 1999, 96, 7714–7718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miletic, G.; Hanson, E.N.; Miletic, V. Brain-derived neurotrophic factor-elicited or sciatic ligation-associated phosphorylation of cyclic AMP response element binding protein in the rat spinal dorsal horn is reduced by block of tyrosine kinase receptors. Neurosci. Lett. 2004, 361, 269–271. [Google Scholar] [CrossRef]
- McMahon, S.B.; Cafferty, W.B. Neurotrophic influences on neuropathic pain. Novartis Found. Symp. 2004, 261, discussion 92–102, 149–154. [Google Scholar]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and modulators of pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef]
- Merighi, A.; Salio, C.; Ghirri, A.; Lossi, L.; Ferrini, F.; Betelli, C.; Bardoni, R. BDNF as a pain modulator. Prog. Neurobiol. 2008, 85, 297–317. [Google Scholar] [CrossRef]
- Li, Y.X.; Xu, Y.; Ju, D.; Lester, H.A.; Davidson, N.; Schuman, E.M. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 10884–10889. [Google Scholar] [CrossRef] [Green Version]
- Michael, G.J.; Averill, S.; Nitkunan, A.; Rattray, M.; Bennett, D.L.; Yan, Q.; Priestley, J.V. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J. Neurosci. 1997, 17, 8476–8490. [Google Scholar] [CrossRef]
- Ha, S.O.; Kim, J.K.; Hong, H.S.; Kim, D.S.; Cho, H.J. Expression of brain-derived neurotrophic factor in rat dorsal root ganglia, spinal cord and gracile nuclei in experimental models of neuropathic pain. Neuroscience 2001, 107, 301–309. [Google Scholar] [CrossRef]
- Pezet, S.; Malcangio, M.; Lever, I.J.; Perkinton, M.S.; Thompson, S.W.; Williams, R.J.; McMahon, S.B. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol. Cell Neurosci. 2002, 21, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zou, S.; Guan, Y.; Ikeda, T.; Tal, M.; Dubner, R.; Ren, K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J. Neurosci. 2002, 22, 6208–6217. [Google Scholar] [CrossRef] [PubMed]
- Renn, C.L.; Lin, L.; Thomas, S.; Dorsey, S.G. Full-length tropomyosin-related kinase B expression in the brainstem in response to persistent inflammatory pain. Neuroreport 2006, 17, 1175–1179. [Google Scholar] [CrossRef]
- Kerr, B.J.; Bradbury, E.J.; Bennett, D.L.; Trivedi, P.M.; Dassan, P.; French, J.; Shelton, D.B.; McMahon, S.B.; Thompson, S.W. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J. Neurosci. 1999, 19, 5138–5148. [Google Scholar] [CrossRef] [Green Version]
- Renn, C.L.; Leitch, C.C.; Lessans, S.; Rhee, P.; McGuire, W.C.; Smith, B.A.; Traub, R.J.; Dorsey, S.G. Brain-derived neurotrophic factor modulates antiretroviral-induced mechanical allodynia in the mouse. J. Neurosci. Res. 2011, 89, 1551–1565. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Ramakrishnan, K.; Croll, S.D.; Siuciak, J.A.; Yu, G.; Young, L.T.; Fahnestock, M. Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav. Neurosci. 2001, 115, 1145–1153. [Google Scholar] [CrossRef]
- Yajima, Y.; Narita, M.; Usui, A.; Kaneko, C.; Miyatake, M.; Narita, M.; Yamaguchi, T.; Tamaki, H.; Wachi, H.; Seyama, Y.; et al. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J. Neurochem. 2005, 93, 584–594. [Google Scholar] [CrossRef]
- Renn, C.L.; Leitch, C.C.; Dorsey, S.G. In vivo evidence that truncated trkB.T1 participates in nociception. Mol. Pain 2009, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Middlemas, D.S.; Lindberg, R.A.; Hunter, T. trkB, a neural receptor protein-tyrosine kinase: Evidence for a full-length and two truncated receptors. Mol. Cell Biol. 1991, 11, 143–153. [Google Scholar] [CrossRef]
- Lee, S.L.; Kim, J.K.; Kim, D.S.; Cho, H.J. Expression of mRNAs encoding full-length and truncated TrkB receptors in rat dorsal root ganglia and spinal cord following peripheral inflammation. Neuroreport 1999, 10, 2847–2851. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, S.G.; Lovering, R.M.; Renn, C.L.; Leitch, C.C.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Pratap, A.; Ott, S.; Sengamalay, N.; et al. Genetic deletion of trkB.T1 increases neuromuscular function. Am. J. Physiol. Cell Physiol. 2012, 302, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squinto, S.P.; Stitt, T.N.; Aldrich, T.H.; Davis, S.; Bianco, S.M.; Radziejewski, C.; Glass, D.J.; Masiakowski, P.; Furth, M.E.; Valenzuela, D.M.; et al. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell 1991, 65, 885–893. [Google Scholar] [CrossRef]
- Cosgaya, J.M.; Chan, J.R.; Shooter, E.M. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 2002, 298, 1245–1248. [Google Scholar] [CrossRef]
- Ohira, K.; Kumanogoh, H.; Sahara, Y.; Homma, K.J.; Hirai, H.; Nakamura, S.; Hayashi, M. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J. Neurosci. 2005, 25, 1343–1353. [Google Scholar] [CrossRef]
- Ohira, K.; Homma, K.J.; Hirai, H.; Nakamura, S.; Hayashi, M. TrkB-T1 regulates the RhoA signaling and actin cytoskeleton in glioma cells. Biochem. Biophys. Res. Commun. 2006, 342, 867–874. [Google Scholar] [CrossRef]
- Holt, L.M.; Hernandez, R.D.; Pacheco, N.L.; Torres Ceja, B.; Hossain, M.; Olsen, M.L. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ohira, K.; Funatsu, N.; Homma, K.J.; Sahara, Y.; Hayashi, M.; Kaneko, T.; Nakamura, S. Truncated TrkB-T1 regulates the morphology of neocortical layer I astrocytes in adult rat brain slices. Eur. J. Neurosci. 2007, 25, 406–416. [Google Scholar] [CrossRef]
- Rose, C.R.; Blum, R.; Pichler, B.; Lepier, A.; Kafitz, K.W.; Konnerth, A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 2003, 426, 74–78. [Google Scholar] [CrossRef]
- Dorsey, S.G.; Renn, C.L.; Carim-Todd, L.; Barrick, C.A.; Bambrick, L.; Krueger, B.K.; Ward, C.W.; Tessarollo, L. In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron 2006, 51, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G. TrkB.T1 contributes to neuropathic pain after spinal cord injury through regulation of cell cycle pathways. J. Neurosci. 2013, 33, 12447–12463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyas, J.J.; O’Driscoll, C.M.; Yu, L.; Coll-Miro, M.; Daugherty, S.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Truncated TrkB.T1-Mediated Astrocyte Dysfunction Contributes to Impaired Motor Function and Neuropathic Pain after Spinal Cord Injury. J. Neurosci. 2017, 37, 3956–3971. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, K.; Zagrebelsky, M.; Berndt-Huch, J.; Polack, M.; Buschler, A.; Sendtner, M.; Korte, M. Neurotrophin receptors TrkB.T1 and p75NTR cooperate in modulating both functional and structural plasticity in mature hippocampal neurons. Eur. J. Neurosci. 2010, 32, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Marin, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Marti, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Vanhecke, E.; Adriaenssens, E.; Verbeke, S.; Meignan, S.; Germain, E.; Berteaux, N.; Nurcombe, V.; Le Bourhis, X.; Hondermarck, H. Brain-derived neurotrophic factor and neurotrophin-4/5 are expressed in breast cancer and can be targeted to inhibit tumor cell survival. Clin. Cancer Res. 2011, 17, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [Green Version]
- Fenner, B.M. Truncated TrkB: Beyond a dominant negative receptor. Cytokine Growth Factor Rev. 2012, 23, 15–24. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Alcantara, S.; Frisen, J.; del Rio, J.A.; Soriano, E.; Barbacid, M.; Silos-Santiago, I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J. Neurosci. 1997, 17, 3623–3633. [Google Scholar] [CrossRef] [Green Version]
- Linnarsson, S.; Willson, C.A.; Ernfors, P. Cell death in regenerating populations of neurons in BDNF mutant mice. Brain Res. Mol. Brain Res. 2000, 75, 61–69. [Google Scholar] [CrossRef]
- Pencea, V.; Bingaman, K.D.; Wiegand, S.J.; Luskin, M.B. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J. Neurosci. 2001, 21, 6706–6717. [Google Scholar] [CrossRef] [Green Version]
- Philippe, J.; Chick, W.L.; Habener, J.F. Multipotential phenotypic expression of genes encoding peptide hormones in rat insulinoma cell lines. J. Clin. Investig. 1987, 79, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantilla, C.B.; Gransee, H.M.; Zhan, W.Z.; Sieck, G.C. Motoneuron BDNF/TrkB signaling enhances functional recovery after cervical spinal cord injury. Exp. Neurol. 2013, 247, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantilla, C.B.; Greising, S.M.; Stowe, J.M.; Zhan, W.Z.; Sieck, G.C. TrkB kinase activity is critical for recovery of respiratory function after cervical spinal cord hemisection. Exp. Neurol. 2014, 261, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Pivac, N.; Nikolac, M.; Nedic, G.; Mustapic, M.; Borovecki, F.; Hajnsek, S.; Presecki, P.; Pavlovic, M.; Mimica, N.; Muck Seler, D. Brain derived neurotrophic factor Val66Met polymorphism and psychotic symptoms in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 356–362. [Google Scholar] [CrossRef]
- Pivac, N.; Kozaric-Kovacic, D.; Grubisic-Ilic, M.; Nedic, G.; Rakos, I.; Nikolac, M.; Blazev, M.; Muck-Seler, D. The association between brain-derived neurotrophic factor Val66Met variants and psychotic symptoms in posttraumatic stress disorder. World J. Biol. Psychiatry 2012, 13, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Buckley, P.F.; Pillai, A.; Howell, K.R. Brain-derived neurotrophic factor: Findings in schizophrenia. Curr. Opin. Psychiatry 2011, 24, 122–127. [Google Scholar] [CrossRef]
- Luberg, K.; Wong, J.; Weickert, C.S.; Timmusk, T. Human TrkB gene: Novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. J. Neurochem. 2010, 113, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Peng, X.; Greene, L.A.; Kaplan, D.R.; Stephens, R.M. Deletion of a conserved juxtamembrane sequence in Trk abolishes NGF-promoted neuritogenesis. Neuron 1995, 15, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Kiris, E.; Wang, T.; Yanpallewar, S.; Dorsey, S.G.; Becker, J.; Bavari, S.; Palko, M.E.; Coppola, V.; Tessarollo, L. TrkA in vivo function is negatively regulated by ubiquitination. J. Neurosci. 2014, 34, 4090–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.; Brigadski, T.; Erdmann, K.S.; Holtmann, B.; Sendtner, M.; Narz, F.; LeBmann, V. Truncated TrkB receptor-induced outgrowth of dendritic filopodia involves the p75 neurotrophin receptor. J. Cell Sci. 2004, 117, 12. [Google Scholar] [CrossRef] [Green Version]
- Ninkina, N.; Adu, J.; Fischer, A.; Pinon, L.G.; Buchman, V.L.; Davies, A.M. Expression and function of TrkB variants in developing sensory neurons. EMBO J. 1996, 15, 6385–6393. [Google Scholar] [CrossRef]
- Frisen, J.; Verge, V.M.; Fried, K.; Risling, M.; Persson, H.; Trotter, J.; Hokfelt, T.; Lindholm, D. Characterization of glial trkB receptors: Differential response to injury in the central and peripheral nervous systems. Proc. Natl. Acad. Sci. USA 1993, 90, 4971–4975. [Google Scholar] [CrossRef] [Green Version]
- Fenner, M.E.; Achim, C.L.; Fenner, B.M. Expression of full-length and truncated trkB in human striatum and substantia nigra neurons: Implications for Parkinson’s disease. J. Mol. Histol. 2014, 45, 349–361. [Google Scholar] [CrossRef]
- Ohira, K.; Shimizu, K.; Hayashi, M. TrkB dimerization during development of the prefrontal cortex of the macaque. J. Neurosci. Res. 2001, 65, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Eide, F.F.; Vining, E.R.; Eide, B.L.; Zang, K.; Wang, X.Y.; Reichardt, L.F. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J. Neurosci. 1996, 16, 3123–3129. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, A.; Koponen, E.; Hoppe, E.; Wong, G.; Castren, E. Truncated trkB.T1 is dominant negative inhibitor of trkB.TK+-mediated cell survival. Biochem. Biophys. Res. Commun. 2001, 280, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Biffo, S.; Offenhauser, N.; Carter, B.D.; Barde, Y.A. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development 1995, 121, 2461–2470. [Google Scholar] [PubMed]
- Fryer, R.H.; Kaplan, D.R.; Kromer, L.F. Truncated trkB receptors on nonneuronal cells inhibit BDNF-induced neurite outgrowth in vitro. Exp. Neurol. 1997, 148, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Curtis, R.; Alterman, A.L.; Lindsay, R.M.; DiStefano, P.S. Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and schwann cells in vitro. Brain Res. 2000, 871, 210–222. [Google Scholar] [CrossRef]
- Baxter, G.T.; Radeke, M.J.; Kuo, R.C.; Makrides, V.; Hinkle, B.; Hoang, R.; Medina-Selby, A.; Coit, D.; Valenzuela, P.; Feinstein, S.C. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J. Neurosci. 1997, 17, 2683–2690. [Google Scholar] [CrossRef]
- Aroeira, R.I.; Sebastiao, A.M.; Valente, C.A. BDNF, via truncated TrkB receptor, modulates GlyT1 and GlyT2 in astrocytes. Glia 2015, 63, 2181–2197. [Google Scholar] [CrossRef]
- Kingsbury, T.J.; Murray, P.D.; Bambrick, L.L.; Krueger, B.K. Ca(2+)-dependent regulation of TrkB expression in neurons. J. Biol. Chem. 2003, 278, 40744–40748. [Google Scholar] [CrossRef] [Green Version]
- Dombert, B.; Balk, S.; Luningschror, P.; Moradi, M.; Sivadasan, R.; Saal-Bauernschubert, L.; Jablonka, S. BDNF/trkB Induction of Calcium Transients through Cav2.2 Calcium Channels in Motoneurons Corresponds to F-actin Assembly and Growth Cone Formation on beta2-Chain Laminin (221). Front. Mol. Neurosci. 2017, 10, 346. [Google Scholar] [CrossRef]
- Fulgenzi, G.; Tomassoni-Ardori, F.; Babini, L.; Becker, J.; Barrick, C.; Puverel, S.; Tessarollo, L. BDNF modulates heart contraction force and long-term homeostasis through truncated TrkB.T1 receptor activation. J. Cell Biol. 2015, 210, 1003–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Conway, D.; Parada, L.F.; Barbacid, M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 1990, 61, 647–656. [Google Scholar] [CrossRef]
- Kryl, D.; Barker, P.A. TTIP is a novel protein that interacts with the truncated T1 TrkB neurotrophin receptor. Biochem. Biophys. Res. Commun. 2000, 279, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Chao, M.V. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. USA 2001, 98, 3555–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, R.; Chen, Z.Y.; Lee, F.S.; Chao, M.V. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J. Neurosci. 2004, 24, 6650–6658. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Lo, D.C. Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat. Neurosci. 2000, 3, 342–349. [Google Scholar] [CrossRef]
- Cheng, A.; Coksaygan, T.; Tang, H.; Khatri, R.; Balice-Gordon, R.J.; Rao, M.S.; Mattson, M.P. Truncated tyrosine kinase B brain-derived neurotrophic factor receptor directs cortical neural stem cells to a glial cell fate by a novel signaling mechanism. J. Neurochem. 2007, 100, 1515–1530. [Google Scholar] [CrossRef]
- Klein, R.; Smeyne, R.J.; Wurst, W.; Long, L.K.; Auerbach, B.A.; Joyner, A.L.; Barbacid, M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 1993, 75, 113–122. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Movsesyan, V.; Ahmed, F.; Cernak, I.; Schinelli, S.; Stoica, B.; Faden, A.I. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. USA 2005, 102, 8333–8338. [Google Scholar] [CrossRef] [Green Version]
- Jeronimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lerias, S.; Caetano, A.P.; Buee-Scherrer, V.; Castren, E.; Valente, C.A.; Blum, D.; Sebastiao, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-beta Peptide is Mediated by Calpain. Cereb. Cortex 2015, 25, 3107–3121. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, G.; Otten, U.; Meier, F.; Baysang, G.; Dimitriades-Schmutz, B.; Muller-Spahn, F.; Savaskan, E. Beta-amyloid modulates tyrosine kinase B receptor expression in SHSY5Y neuroblastoma cells: Influence of the antioxidant melatonin. Neuroscience 2003, 120, 659–665. [Google Scholar] [CrossRef]
- Ansaloni, S.; Leung, B.P.; Sebastian, N.P.; Samudralwar, R.; Gadaleta, M.; Saunders, A.J. TrkB Isoforms Differentially Affect AICD Production through Their Intracellular Functional Domains. Int. J. Alzheimers Dis. 2011, 2011, 729382. [Google Scholar] [CrossRef] [Green Version]
- Real, C.C.; Ferreira, A.F.; Chaves-Kirsten, G.P.; Torrao, A.S.; Pires, R.S.; Britto, L.R. BDNF receptor blockade hinders the beneficial effects of exercise in a rat model of Parkinson’s disease. Neuroscience 2013, 237, 118–129. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Shruthi, S.; Sumitha, R.; Varghese, A.M.; Ashok, S.; Chandrasekhar Sagar, B.K.; Sathyaprabha, T.N.; Nalini, A.; Kramer, B.W.; Raju, T.R.; Vijayalakshmi, K.; et al. Brain-Derived Neurotrophic Factor Facilitates Functional Recovery from ALS-Cerebral Spinal Fluid-Induced Neurodegenerative Changes in the NSC-34 Motor Neuron Cell Line. Neurodegener. Dis. 2017, 17, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutoh, T.; Sobue, G.; Hamano, T.; Kuriyama, M.; Hirayama, M.; Yamamoto, M.; Mitsuma, T. Decreased phosphorylation levels of TrkB neurotrophin receptor in the spinal cords from patients with amyotrophic lateral sclerosis. Neurochem. Res. 2000, 25, 239–245. [Google Scholar] [CrossRef]
- Quarta, E.; Fulgenzi, G.; Bravi, R.; Cohen, E.J.; Yanpallewar, S.; Tessarollo, L.; Minciacchi, D. Deletion of the endogenous TrkB.T1 receptor isoform restores the number of hippocampal CA1 parvalbumin-positive neurons and rescues long-term potentiation in pre-symptomatic mSOD1(G93A) ALS mice. Mol. Cell Neurosci. 2018, 89, 33–41. [Google Scholar] [CrossRef]
- Yanpallewar, S.U.; Barrick, C.A.; Buckley, H.; Becker, J.; Tessarollo, L. Deletion of the BDNF truncated receptor TrkB. T1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. 2012, 7, 39946. [Google Scholar]
- Liebl, D.J.; Huang, W.; Young, W.; Parada, L.F. Regulation of Trk receptors following contusion of the rat spinal cord. Exp. Neurol. 2001, 167, 15–26. [Google Scholar] [CrossRef]
- Widenfalk, J.; Lundstromer, K.; Jubran, M.; Brene, S.; Olson, L. Neurotrophic factors and receptors in the immature and adult spinal cord after mechanical injury or kainic acid. J. Neurosci. 2001, 21, 3457–3475. [Google Scholar] [CrossRef] [PubMed]
- King, V.R.; Bradbury, E.J.; McMahon, S.B.; Priestley, J.V. Changes in truncated trkB and p75 receptor expression in the rat spinal cord following spinal cord hemisection and spinal cord hemisection plus neurotrophin treatment. Exp. Neurol. 2000, 165, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Garraway, S.M.; Mendell, L.M. Spinal cord transection enhances afferent-evoked inhibition in lamina II neurons and abolishes BDNF-induced facilitation of their sensory input. J. Neurotrauma 2007, 24, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Belluardo, N.; Salin, T.; Dell’Albani, P.; Mudo, G.; Corsaro, M.; Jiang, X.H.; Timmusk, T.; Condorelli, D.F. Neurotoxic injury in rat hippocampus differentially affects multiple trkB and trkC transcripts. Neurosci. Lett. 1995, 196, 1–4. [Google Scholar] [CrossRef]
- Wong, J.Y.; Liberatore, G.T.; Donnan, G.A.; Howells, D.W. Expression of brain-derived neurotrophic factor and TrkB neurotrophin receptors after striatal injury in the mouse. Exp. Neurol. 1997, 148, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Rudge, J.S.; Mather, P.E.; Pasnikowski, E.M.; Cai, N.; Corcoran, T.; Acheson, A.; Anderson, K.; Lindsay, R.M.; Wiegand, S.J. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp. Neurol. 1998, 149, 398–410. [Google Scholar] [CrossRef]
- Venero, J.L.; Hefti, F. Regionally specific induction of BDNF and truncated trkB.T1 receptors in the hippocampal formation after intraseptal injection of kainic acid. Brain Res. 1998, 790, 270–277. [Google Scholar] [CrossRef]
- Ferrer, I.; Krupinski, J.; Goutan, E.; Marti, E.; Ambrosio, S.; Arenas, E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta. Neuropathol. 2001, 101, 229–238. [Google Scholar] [CrossRef]
- Mamounas, L.A.; Altar, C.A.; Blue, M.E.; Kaplan, D.R.; Tessarollo, L.; Lyons, W.E. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J. Neurosci. 2000, 20, 771–782. [Google Scholar] [CrossRef]
- Saarelainen, T.; Lukkarinen, J.A.; Koponen, S.; Grohn, O.H.; Jolkkonen, J.; Koponen, E.; Haapasalo, A.; Alhonen, L.; Wong, G.; Koistinaho, J.; et al. Transgenic mice overexpressing truncated trkB neurotrophin receptors in neurons show increased susceptibility to cortical injury after focal cerebral ischemia. Mol. Cell Neurosci. 2000, 16, 87–96. [Google Scholar] [CrossRef]
- Vidaurre, O.G.; Gascon, S.; Deogracias, R.; Sobrado, M.; Cuadrado, E.; Montaner, J.; Rodriguez-Pena, A.; Diaz-Guerra, M. Imbalance of neurotrophin receptor isoforms TrkB-FL/TrkB-T1 induces neuronal death in excitotoxicity. Cell Death Dis. 2012, 3, e256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbeck, J.A.; Methner, A. Translational downregulation of the noncatalytic growth factor receptor TrkB.T1 by ischemic preconditioning of primary neurons. Gene Expr. 2005, 12, 99–106. [Google Scholar] [CrossRef]
- Gomes, J.R.; Costa, J.T.; Melo, C.V.; Felizzi, F.; Monteiro, P.; Pinto, M.J.; Inacio, A.R.; Wieloch, T.; Almeida, R.D.; Graos, M.; et al. Excitotoxicity downregulates TrkB.FL signaling and upregulates the neuroprotective truncated TrkB receptors in cultured hippocampal and striatal neurons. J. Neurosci. 2012, 32, 4610–4622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, W.D. Long-term potentiation in spinothalamic neurons. Brain Res. Brain Res. Rev. 2002, 40, 202–214. [Google Scholar] [CrossRef]
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836. [Google Scholar] [CrossRef] [Green Version]
- Maratou, K.; Wallace, V.C.; Hasnie, F.S.; Okuse, K.; Hosseini, R.; Jina, N.; Blackbeard, J.; Pheby, T.; Orengo, C.; Dickenson, A.H.; et al. Comparison of dorsal root ganglion gene expression in rat models of traumatic and HIV-associated neuropathic pain. Eur. J. Pain 2009, 13, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Boudes, M.; Menigoz, A. Non-neuronal BDNF, a key player in development of central sensitization and neuropathic pain. J. Physiol. 2009, 587, 2111–2112. [Google Scholar] [CrossRef]
- Carlton, S.M.; Du, J.; Tan, H.Y.; Nesic, O.; Hargett, G.L.; Bopp, A.C.; Yamani, A.; Lin, Q.; Willis, W.D.; Hulsebosch, C.E. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 2009, 147, 265–276. [Google Scholar] [CrossRef]
- Hains, B.C.; Klein, J.P.; Saab, C.Y.; Craner, M.J.; Black, J.A.; Waxman, S.G. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 2003, 23, 8881–8892. [Google Scholar] [CrossRef] [Green Version]
- Drew, G.M.; Siddall, P.J.; Duggan, A.W. Responses of spinal neurones to cutaneous and dorsal root stimuli in rats with mechanical allodynia after contusive spinal cord injury. Brain Res. 2001, 893, 59–69. [Google Scholar] [CrossRef]
- Detloff, M.R.; Fisher, L.C.; McGaughy, V.; Longbrake, E.E.; Popovich, P.G.; Basso, D.M. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp. Neurol. 2008, 212, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.J.; Ji, R.R. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010, 7, 482–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, K.R.; Stoica, B.A.; Fricke, S.; Di Giovanni, S.; Faden, A.I. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain 2007, 130, 2977–2992. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Stoica, B.A.; Faden, A.I. Cell cycle activation and spinal cord injury. Neurotherapeutics 2011, 8, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, M.; Kohro, Y.; Yano, T.; Tsujikawa, T.; Kitano, J.; Tozaki-Saitoh, H.; Koyanagi, S.; Ohdo, S.; Ji, R.R.; Salter, M.W.; et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain 2011, 134, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; He, Y.; Zhang, Y.; Li, Y.; Li, Y.; Han, Y.; Zhu, H.; Wang, Y. Activation of cyclin-dependent kinase 5 (Cdk5) in primary sensory and dorsal horn neurons by peripheral inflammation contributes to heat hyperalgesia. Pain 2007, 127, 109–120. [Google Scholar] [CrossRef]
- Wang, C.H.; Chou, W.Y.; Hung, K.S.; Jawan, B.; Lu, C.N.; Liu, J.K.; Hung, Y.P.; Lee, T.H. Intrathecal administration of roscovitine inhibits Cdk5 activity and attenuates formalin-induced nociceptive response in rats. Acta Pharmacol. Sin. 2005, 26, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, Z.; Zhu, X.; Renn, C.L.; Dorsey, S.G.; Faden, A.I. Cell cycle inhibition limits development and maintenance of neuropathic pain following spinal cord injury. Pain 2016, 157, 488–503. [Google Scholar] [CrossRef]
- Wu, J.; Raver, C.; Piao, C.; Keller, A.; Faden, A.I. Cell cycle activation contributes to increased neuronal activity in the posterior thalamic nucleus and associated chronic hyperesthesia after rat spinal cord contusion. Neurotherapeutics 2013, 10, 520–538. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sabirzhanov, B.; Stoica, B.A.; Lipinski, M.M.; Zhao, Z.; Zhao, S.; Ward, N.; Yang, D.; Faden, A.I. Ablation of the transcription factors E2F1-2 limits neuroinflammation and associated neurological deficits after contusive spinal cord injury. Cell Cycle 2015, 14, 3698–3712. [Google Scholar] [CrossRef] [Green Version]
- Hains, B.C.; Waxman, S.G. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 2006, 26, 4308–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwak, Y.S.; Kang, J.; Unabia, G.C.; Hulsebosch, C.E. Spatial and temporal activation of spinal glial cells: Role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp. Neurol. 2012, 234, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, E.T. Neuroinflammatory contributions to pain after SCI: Roles for central glial mechanisms and nociceptor-mediated host defense. Exp. Neurol. 2014, 258, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. 1), 10–28. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef]
- Filous, A.R.; Silver, J. “Targeting astrocytes in CNS injury and disease: A translational research approach”. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, T.; Matyas, J.J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells 2020, 9, 1194. https://doi.org/10.3390/cells9051194
Cao T, Matyas JJ, Renn CL, Faden AI, Dorsey SG, Wu J. Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells. 2020; 9(5):1194. https://doi.org/10.3390/cells9051194
Chicago/Turabian StyleCao, Tuoxin, Jessica J. Matyas, Cynthia L. Renn, Alan I. Faden, Susan G. Dorsey, and Junfang Wu. 2020. "Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain" Cells 9, no. 5: 1194. https://doi.org/10.3390/cells9051194
APA StyleCao, T., Matyas, J. J., Renn, C. L., Faden, A. I., Dorsey, S. G., & Wu, J. (2020). Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells, 9(5), 1194. https://doi.org/10.3390/cells9051194