Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α


{kind=link}
{kind=link}
Abstract
:1. Alzheimer Disease: A Dementing Illness
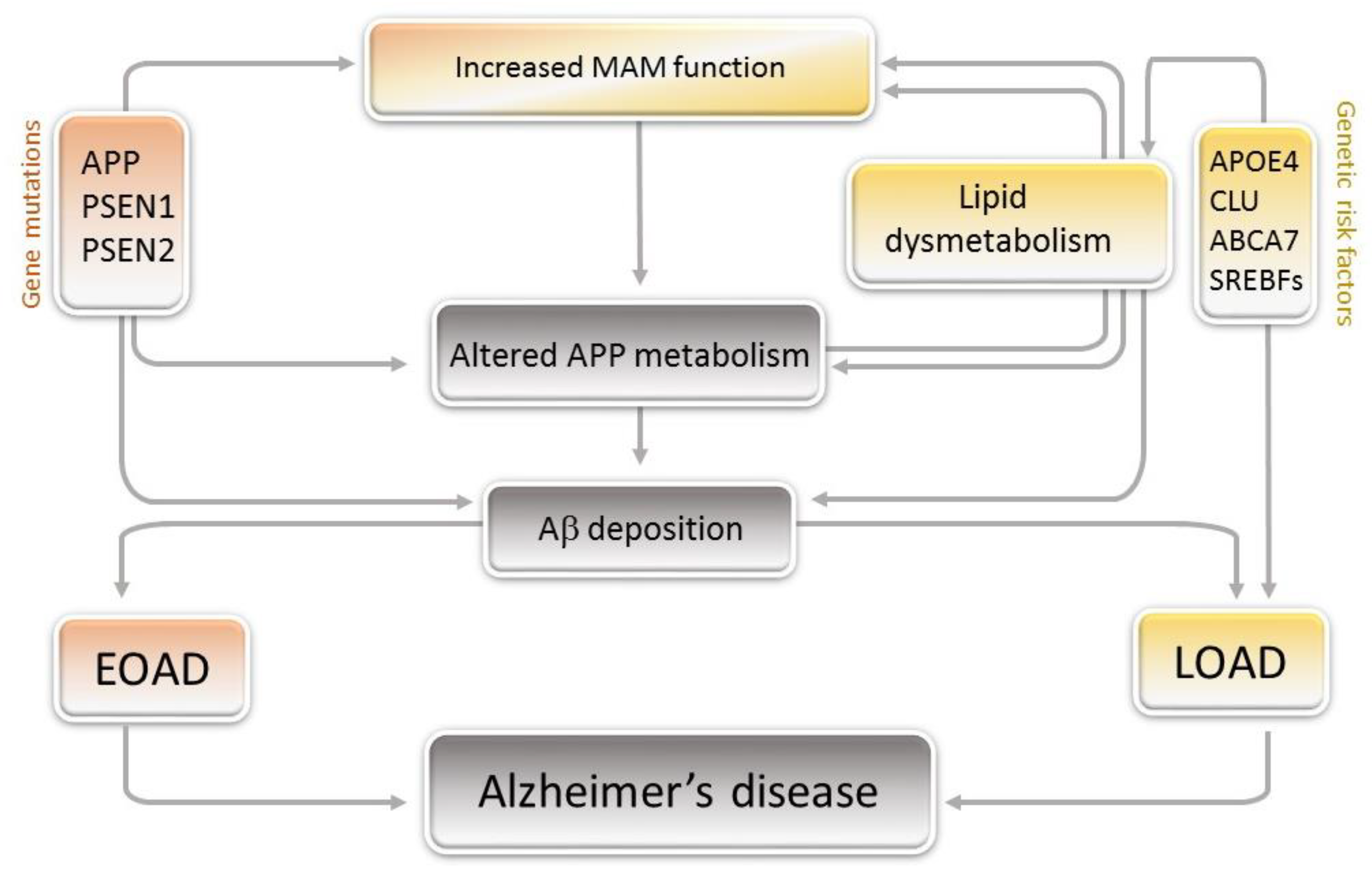
2. Linking Lipids to Alzheimer’s Disease
3. Lipids in Alzheimer Disease: Involvement of Fatty Acids in Cognitive Function
4. RXRs, LXRs and PPARs Nuclear Receptors in AD
4.1. Nuclear Receptors
4.2. RXRs
4.3. LXRs
4.4. PPARs
5. PPARs in Alzheimer’s Disease Therapy: The Promising Role of PPARα
5.1. PPARγ and PPARβ/δ in AD
5.2. PPARα in AD
5.2.1. PPARα Function in Brain Energy Supply
5.2.2. PPARα and Cognitive Function
5.2.3. Potential Link between PPARα and AD
5.2.4. PPARα Ligands and AD
5.2.5. PPARα and Sex
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Disease International. World Alzheimer Report 2019, Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Feldman, H.H.; Jacova, C.; Robillard, A.; Garcia, A.; Chow, T.; Borrie, M.; Schipper, H.M.; Blair, M.; Kertesz, A.; Chertkow, H. Diagnosis and treatment of dementia: 2. Diagnosis. Can. Med. Assoc. J. 2008, 178, 825–836. [Google Scholar] [CrossRef] [Green Version]
- Kuslansky, G.; Katz, M.; Verghese, J.; Hall, C.B.; Lapuerta, P.; LaRuffa, G.; Lipton, R.B. Detecting dementia with the Hopkins Verbal Learning Test and the Mini-Mental State Examination. Arch. Clin. Neuropsychol. 2004, 19, 89–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnetti, L.; Lanari, A.; Silvestrelli, G.; Saggese, E.; Reboldi, P. Diagnosing prodromal Alzheimer’s disease: Role of CSF biochemical markers. Mech. Ageing Dev. 2006, 127, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaccarino, L.; Sala, A.; Caminiti, S.; Perani, D. The emerging role of PET imaging in dementia [version 1; peer review: 3 approved]. F1000 Res. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Doré, V.; Burnham, S.C.; Masters, C.L.; Rowe, C.C. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 2018, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Bergmans, B.A.; De Strooper, B. γ-secretases: From cell biology to therapeutic strategies. Lancet Neurol. 2010, 9, 215–226. [Google Scholar] [CrossRef]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investig. 2019, 99, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Gedalya, T.; Moll, L.; Bejerano-Sagie, M.; Frere, S.; Cabral, W.A.; Friedmann-Morvinski, D.; Slutsky, I.; Burstyn-Cohen, T.; Marini, J.C.; Cohen, E. Alzheimer’s disease-causing proline substitutions lead to presenilin 1 aggregation and malfunction. EMBO J. 2015, 34, 2820–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J. Presenilin-1 Knockin Mice Reveal Loss-of-Function Mechanism for Familial Alzheimer’s Disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szaruga, M.; Veugelen, S.; Benurwar, M.; Lismont, S.; Sepulveda-Falla, D.; Lleo, A.; Ryan, N.S.; Lashley, T.; Fox, N.C.; Murayama, S.; et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J. Exp. Med. 2015, 212, 2003–2013. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther. 2014, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Tseng, B.P.; Rydel, R.E.; Podlisny, M.B.; Selkoe, D.J. The Oligomerization of Amyloid β-Protein Begins Intracellularly in Cells Derived from Human Brain. Biochemistry 2000, 39, 10831–10839. [Google Scholar] [CrossRef]
- Chen, Y.-R.; Glabe, C.G. Distinct Early Folding and Aggregation Properties of Alzheimer Amyloid-β Peptides Aβ40 and Aβ42: STABLE TRIMER OR TETRAMER FORMATION BY Aβ42. J. Biol. Chem. 2006, 281, 24414–24422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesné, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.; Paulsson, J.F.; Blinder, P.; Burstyn-Cohen, T.; Du, D.; Estepa, G.; Adame, A.; Pham, H.M.; Holzenberger, M.; Kelly, J.W.; et al. Reduced IGF-1 Signaling Delays Age-Associated Proteotoxicity in Mice. Cell 2009, 139, 1157–1169. [Google Scholar] [CrossRef] [Green Version]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid β-Protein Dimers Rapidly Form Stable Synaptotoxic Protofibrils. J. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.-H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef]
- Eisai. Eisai And Biogen To Discontinue Phase III Clinical Studies Of BACE Inhibitor Elenbecestat In Early Alzheimer’s Disease. Available online: http://eisai.mediaroom.com/2019-09-13-Eisai-And-Biogen-To-Discontinue-Phase-III-Clinical-Studies-Of-BACE-Inhibitor-Elenbecestat-In-Early-Alzheimers-Disease (accessed on 1 April 2020).
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target 2019, 4, 29. [Google Scholar] [CrossRef]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s Disease: An Update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Shimano, H.; Shimomura, I.; Hammer, R.E.; Herz, J.; Goldstein, J.L.; Brown, M.S.; Horton, J.D. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J. Clin. Investig. 1997, 100, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Bang, S.; McMullen, M.F.; Kazi, H.; Talbot, K.; Ho, M.-X.; Carlson, G.; Arnold, S.E.; Ong, W.-Y.; Kim, S.F. Neuronal Activity-Induced Sterol Regulatory Element Binding Protein-1 (SREBP1) is Disrupted in Dysbindin-Null Mice—Potential Link to Cognitive Impairment in Schizophrenia. Mol. Neurobiol. 2017, 54, 1699–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, C.; Julien, C.; Frappier, J.; Miron, J.; Théroux, L.; Dea, D.; Breitner, J.C.S.; Poirier, J. Alterations in cholesterol metabolism–related genes in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 66, e181–e189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steen, V.M.; Skrede, S.; Polushina, T.; López, M.; Andreassen, O.A.; Fernø, J.; Hellard, S.L. Genetic evidence for a role of the SREBP transcription system and lipid biosynthesis in schizophrenia and antipsychotic treatment. Eur. Neuropsychopharmacol. 2017, 27, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, F.; Shen, K.; Wang, W.; Siedlak, S.L.; Lee, H.-g.; Phelix, C.F.; Perry, G.; Shen, L.; Tang, B.; et al. The sterol regulatory element-binding protein 2 is dysregulated by tau alterations in Alzheimer disease. Brain Pathol. 2019, 29, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Pierrot, N.; Tyteca, D.; D’auria, L.; Dewachter, I.; Gailly, P.; Hendrickx, A.; Tasiaux, B.; Haylani, L.E.; Muls, N.; N’Kuli, F.; et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. Embo Mol. Med. 2013, 5, 608–625. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Schneider, J.A.; Tangney, C.; Tremblay-Mercier, J.; Fortier, M.; Bennett, D.A.; Morris, M.C. Plasma and Brain Fatty Acid Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 29, 691–697. [Google Scholar] [CrossRef]
- Joffre, C.; Nadjar, A.; Lebbadi, M.; Calon, F.; Laye, S. n-3 LCPUFA improves cognition: The young, the old and the sick. ProstaglandinsLeukot. Essent. Fat. Acids 2014, 91, 1–20. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Pappolla, M.; Pelaez, R.P.; Bazan, N.G. Alzheimer’s Disease—A Dysfunction in Cholesterol and Lipid Metabolism. Cell. Mol. Neurobiol. 2005, 25, 475–483. [Google Scholar] [CrossRef]
- Proitsi, P.; Kim, M.; Whiley, L.; Pritchard, M.; Leung, R.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; et al. Plasma lipidomics analysis finds long chain cholesteryl esters to be associated with Alzheimer’s disease. Transl. Psychiatry 2015, 5, e494. [Google Scholar] [CrossRef] [Green Version]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [Green Version]
- Amtul, Z.; Uhrig, M.; Wang, L.; Rozmahel, R.F.; Beyreuther, K. Detrimental effects of arachidonic acid and its metabolites in cellular and mouse models of Alzheimer’s disease: Structural insight. Neurobiol. Aging 2012, 33, e821–e831. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Fernández, A.; Martínez, L.; Fernández-Checa, J.C.; Colell, A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Aβ accumulation and tau pathology underlying Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3460–3476. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Kuchenbecker, J.; Grösgen, S.; Burg, V.K.; Hundsdörfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic Acid Reduces Amyloid β Production via Multiple Pleiotropic Mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A.; Hillard, C.J.; Spector, A.A.; Watkins, P.A. Brain Uptake and Utilization of Fatty Acids, Lipids and Lipoproteins: Application to Neurological Disorders. J. Mol. Neurosci. 2007, 33, 2–11. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Michaelson, D.M.; Hartmann, T. Omega-3 fatty acids, lipids, and apoE lipidation in Alzheimer’s disease: A rationale for multi-nutrient dementia prevention. J. Lipid Res. 2017, 58, 2083–2101. [Google Scholar] [CrossRef] [Green Version]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.D. A new mechanism of nervous system plasticity: Activity-dependent myelination. Nat. Rev. Neurosci. 2015, 16, 756–767. [Google Scholar] [CrossRef]
- Duncan, A.L.; Song, W.; Sansom, M.S.P. Lipid-Dependent Regulation of Ion Channels and G Protein–Coupled Receptors: Insights from Structures and Simulations. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 31–50. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Vásquez, V. How lipids contribute to ion channel function, a fat perspective on direct and indirect interactions. Curr. Opin. Struct. Biol. 2018, 51, 92–98. [Google Scholar] [CrossRef]
- Munro, S. Lipid Rafts: Elusive or Illusive? Cell 2003, 115, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Martín, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marín, R.; Ferrer, I.; Díaz, M. Lipid Alterations in Lipid Rafts from Alzheimer’s Disease Human Brain Cortex. J. Alzheimer’s Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Raquel, M.; Noemí, F.; Cecilia, F.-E.; Ana, C.-A.; Deiene, R.-B.; David, Q.-A.; Fátima, M.-H.; Mario, D. Lipid Raft Alterations in Aged-Associated Neuropathologies. Curr. Alzheimer Res. 2016, 13, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Hicks, D.; Nalivaeva, N.; Turner, A. Lipid Rafts and Alzheimer’s Disease: Protein-Lipid Interactions and Perturbation of Signaling. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Dart, C. SYMPOSIUM REVIEW: Lipid microdomains and the regulation of ion channel function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Coburger, I.; Hoefgen, S.; Than, M.E. The structural biology of the amyloid precursor protein APP—A complex puzzle reveals its multi-domain architecture. Biol. Chem. 2014, 395, 485–498. [Google Scholar] [CrossRef]
- Van der Kant, R.; Goldstein, L.S.B. Cellular Functions of the Amyloid Precursor Protein from Development to Dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef] [Green Version]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Ludewig, S.; Korte, M. Novel Insights into the Physiological Function of the APP (Gene) Family and Its Proteolytic Fragments in Synaptic Plasticity. Front. Mol. Neurosci. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.W.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Hefter, D.; Ludewig, S.; Draguhn, A.; Korte, M. Amyloid, APP, and Electrical Activity of the Brain. Neuroscientist 2020, 26, 231–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DelBove, C.E.; Strothman, C.E.; Lazarenko, R.M.; Huang, H.; Sanders, C.R.; Zhang, Q. Reciprocal modulation between amyloid precursor protein and synaptic membrane cholesterol revealed by live cell imaging. Neurobiol. Dis. 2019, 127, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. Embo J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. Embo J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. Faseb J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Srivastav, R.K.; Ansari, T.M.; Prasad, M.; Vishwakarma, V.K.; Bagga, P.; Ahsan, F. Caveolins; An Assailant or An Ally of Various Cellular Disorders. Drug Res (Stuttg) 2019, 69, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, N.B. Membrane dynamics, cholesterol homeostasis, and Alzheimer’s disease. J. Lipid Res. 2003, 44, 2019–2029. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, S.B.; Dea, D.; Poirier, J. Increased caveolin-1 expression in Alzheimer’s disease brain. Neurobiol. Aging 2004, 25, 753–759. [Google Scholar] [CrossRef]
- Alsaqati, M.; Thomas, R.S.; Kidd, E.J. Proteins Involved in Endocytosis Are Upregulated by Ageing in the Normal Human Brain: Implications for the Development of Alzheimer’s Disease. J. Gerontol.: Ser. A 2017, 73, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Cameron, P.L.; Ruffin, J.W.; Bollag, R.; Rasmussen, H.; Cameron, R.S. Identification of Caveolin and Caveolin-Related Proteins in the Brain. J. Neurosci. 1997, 17, 9520–9535. [Google Scholar] [CrossRef]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of Caveolin-1 Accelerates Neurodegeneration and Aging. PLoS ONE 2010, 5, e15697. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Ke, D.-S.; Chen, J.-Y. Essential Fatty Acids and Human Brain. Acta Neurol. Taiwanica 2009, 18, 231–241. [Google Scholar]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.A. Polyunsaturated fatty acid synthesis and release by brain-derived cells in vitro. J. Mol. Neurosci. 2001, 16, 195–200. [Google Scholar] [CrossRef]
- Luchtman, D.W.; Song, C. Cognitive enhancement by omega-3 fatty acids from child-hood to old age: Findings from animal and clinical studies. Neuropharmacology 2013, 64, 550–565. [Google Scholar] [CrossRef]
- Hussain, G.; Schmitt, F.; Loeffler, J.-P.; Gonzalez De Aguilar, J. Fatting the brain: A brief of recent research. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef]
- Schaefer, E.J.; Bongard, V.; Beiser, A.S.; Lamon-Fava, S.; Robins, S.J.; Au, R.; Tucker, K.L.; Kyle, D.J.; Wilson, P.W.F.; Wolf, P.A. Plasma Phosphatidylcholine Docosahexaenoic Acid Content and Risk of Dementia and Alzheimer Disease: The Framingham Heart Study. Arch. Neurol. 2006, 63, 1545–1550. [Google Scholar] [CrossRef]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol.-Ren. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R.; Weiner, M.; et al. Docosahexaenoic Acid Supplementation and Cognitive Decline in Alzheimer Disease: A Randomized Trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef]
- Mazereeuw, G.; Lanctôt, K.L.; Chau, S.A.; Swardfager, W.; Herrmann, N. Effects of omega-3 fatty acids on cognitive performance: A meta-analysis. Neurobiol. Aging 2012, 33, e1417–e1429. [Google Scholar] [CrossRef]
- Layé, S. Polyunsaturated fatty acids, neuroinflammation and well being. ProstaglandinsLeukot. Essent. Fat. Acids (Plefa) 2010, 82, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Féart, C.; Peuchant, E.; Letenneur, L.; Samieri, C.; Montagnier, D.; Fourrier-Reglat, A.; Barberger-Gateau, P. Plasma eicosapentaenoic acid is inversely associated with severity of depressive symptomatology in the elderly: Data from the Bordeaux sample of the Three-City Study. Am. J. Clin. Nutr. 2008, 87, 1156–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund-Levi, Y.; Eriksdotter-Jönhagen, M.; Cederholm, T.; Basun, H.; Faxén-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.-O.; Palmblad, J. ω-3 Fatty Acid Treatment in 174 Patients With Mild to Moderate Alzheimer Disease: OmegAD Study: A Randomized Double-blind Trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, F.; Lim, G.P.; Yang, F.; Morihara, T.; Teter, B.; Ubeda, O.; Rostaing, P.; Triller, A.; Salem, N.; Ashe, K.H.; et al. Docosahexaenoic Acid Protects from Dendritic Pathology in an Alzheimer’s Disease Mouse Model. Neuron 2004, 43, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Calon, F.; Cole, G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studies. ProstaglandinsLeukot. Essent. Fat. Acids 2007, 77, 287–293. [Google Scholar] [CrossRef]
- Connor, S.; Tenorio, G.; Clandinin, M.T.; Sauvé, Y. DHA supplementation enhances high-frequency, stimulation-induced synaptic transmission in mouse hippocampus. Appl. Physiol. Nutr. Metab. 2012, 37, 880–887. [Google Scholar] [CrossRef]
- Labrousse, V.F.; Nadjar, A.; Joffre, C.; Costes, L.; Aubert, A.; Grégoire, S.; Bretillon, L.; Layé, S. Short-Term Long Chain Omega3 Diet Protects from Neuroinflammatory Processes and Memory Impairment in Aged Mice. PLoS ONE 2012, 7, e36861. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hossain, S.; Shimada, T.; Sugioka, K.; Yamasaki, H.; Fujii, Y.; Ishibashi, Y.; Oka, J.-I.; Shido, O. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. J. Neurochem. 2002, 81, 1084–1091. [Google Scholar] [CrossRef]
- Cole, G.M.; Frautschy, S.A. Docosahexaenoic Acid Protects from Amyloid and Dendritic Pathology in an Alzheimer’s Disease Mouse Model. Nutr. Health 2006, 18, 249–259. [Google Scholar] [CrossRef]
- Kavraal, S.; Oncu, S.K.; Bitiktas, S.; Artis, A.S.; Dolu, N.; Gunes, T.; Suer, C. Maternal intake of Omega-3 essential fatty acids improves long term potentiation in the dentate gyrus and Morris water maze performance in rats. Brain Res. 2012, 1482, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef] [PubMed]
- Moutinho, M.; Landreth, G.E. Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J. Lipid Res. 2017, 58, 1937–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolezzi, J.M.; Santos, M.J.; Bastías-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. PPARs in the central nervous system: Roles in neurodegeneration and neuroinflammation. Biol. Rev. 2017, 92, 2046–2069. [Google Scholar] [CrossRef]
- Wolf, G. Is 9-Cis-Retinoic Acid the Endogenous Ligand for the Retinoic Acid-X Receptor? Nutr. Rev. 2006, 64, 532–538. [Google Scholar] [CrossRef]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Chitranshi, N.; Dheer, Y.; Kumar, S.; Graham, S.L.; Gupta, V. Molecular docking, dynamics, and pharmacology studies on bexarotene as an agonist of ligand-activated transcription factors, retinoid X receptors. J. Cell. Biochem. 2019, 120, 11745–11760. [Google Scholar] [CrossRef]
- Zhang, C.; Duvic, M. Treatment of cutaneous T-cell lymphoma with retinoids. Dermatol. Ther. 2006, 19, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Lee, C.Y.D.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE Promotes the Proteolytic Degradation of Aβ. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, M.M.; Malm, T.; Lamb, R.; Jay, T.R.; Neilson, L.; Casali, B.; Medarametla, L.; Landreth, G.E. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci. Rep. 2017, 7, 42270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrot, N.; Ris, L.; Stancu, I.-C.; Doshina, A.; Ribeiro, F.; Tyteca, D.; Baugé, E.; Lalloyer, F.; Malong, L.; Schakman, O.; et al. Sex-regulated gene dosage effect of PPARα on synaptic plasticity. Life Sci. Alliance 2019, 2, e201800262. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; Jay, T.; Goduni, E.; Quigley, C.; Mariani, M.M.; Malm, T.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Nuclear Receptors License Phagocytosis by Trem2+ Myeloid Cells in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2015, 35, 6532–6543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARγ/RXRα-Induced and CD36-Mediated Microglial Amyloid-β Phagocytosis Results in Cognitive Improvement in Amyloid Precursor Protein/Presenilin 1 Mice. J. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Lin, S.; Beyer, T.P.; Zhang, Y.; Wu, X.; Bales, K.R.; DeMattos, R.B.; May, P.C.; Li, S.D.; Jiang, X.-C.; et al. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J. Neurochem. 2004, 88, 623–634. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, Y.; Liu, C.-C.; Shinohara, M.; Nielsen, H.M.; Dong, Q.; Kanekiyo, T.; Bu, G. Retinoic Acid Isomers Facilitate Apolipoprotein E Production and Lipidation in Astrocytes through the Retinoid X Receptor/Retinoic Acid Receptor Pathway. J. Biol. Chem. 2014, 289, 11282–11292. [Google Scholar] [CrossRef] [Green Version]
- Pierrot, N.; Lhommel, R.; Quenon, L.; Hanseeuw, B.; Dricot, L.; Sindic, C.; Maloteaux, J.-M.; Octavea, J.-N.; Ivanoiu, A. Targretin Improves Cognitive and Biological Markers in a Patient with Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 49, 271–276. [Google Scholar] [CrossRef]
- Ghosal, K.; Haag, M.; Verghese, P.B.; West, T.; Veenstra, T.; Braunstein, J.B.; Bateman, R.J.; Holtzman, D.M.; Landreth, G.E. A randomized controlled study to evaluate the effect of bexarotene on amyloid-β and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement (N Y) 2016, 2, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L.; Zhong, K.; Kinney, J.W.; Heaney, C.; Moll-Tudla, J.; Joshi, A.; Pontecorvo, M.; Devous, M.; Tang, A.; Bena, J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2016, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Schroepfer, J. Oxysterols: Modulators of Cholesterol Metabolism and Other Processes. Physiol. Rev. 2000, 80, 361–554. [Google Scholar] [CrossRef]
- Ma, L.; Nelson, E.R. Oxysterols and nuclear receptors. Mol. Cell. Endocrinol. 2019, 484, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Kaplan, R.; Menke, J.G.; MacNaul, K.; Chen, Y.; Sparrow, C.P.; Zhou, G.; Wright, S.D.; Cai, T.-Q. Dual Mechanisms of ABCA1 Regulation by Geranylgeranyl Pyrophosphate. J. Biol. Chem. 2001, 276, 48702–48708. [Google Scholar] [CrossRef] [Green Version]
- Langmann, T.; Liebisch, G.; Moehle, C.; Schifferer, R.; Dayoub, R.; Heiduczek, S.; Grandl, M.; Dada, A.; Schmitz, G. Gene expression profiling identifies retinoids as potent inducers of macrophage lipid efflux. Biochim. Biophys. Acta (Bba)-Mol. Basis Dis. 2005, 1740, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.; Tu, H.; Shan, B.; Luk, A.; DeBose-Boyd, R.A.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci. USA 2001, 98, 6027–6032. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Shimano, H.; Yahagi, N.; Ide, T.; Amemiya-Kudo, M.; Matsuzaka, T.; Nakakuki, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Polyunsaturated Fatty Acids Suppress Sterol Regulatory Element-binding Protein 1c Promoter Activity by Inhibition of Liver X Receptor (LXR) Binding to LXR Response Elements. J. Biol. Chem. 2002, 277, 1705–1711. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.K.; Rao, M.S. Xenobiotic-Induced Peroxisome Proliferation: Role of Tissue Specificity and Species Differences in Response in the Evaluation of the Implications for Human Health; Springer: Berlin/Heidelberg, Germany, 1987; pp. 43–53. [Google Scholar]
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of mRNAs of Peroxisome Proliferator–Activated Receptors and Liver X Receptor-α in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, P.; Arrabal, S.; Vargas, A.; Blanco Calvo, E.; Serrano, A.; Pavon, F.J.; Rodriguez de Fonseca, F.; Suarez, J. Localization of peroxisome proliferator-activated receptor alpha (PPARα) and N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) in cells expressing the Ca2+-binding proteins calbindin, calretinin, and parvalbumin in the adult rat hippocampus. Front. Neuroanat. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Moreno, S.; Farioli-Vecchioli, S.; Cerù, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid x receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Gofflot, F.; Chartoire, N.; Vasseur, L.; Heikkinen, S.; Dembele, D.; Le Merrer, J.; Auwerx, J. Systematic Gene Expression Mapping Clusters Nuclear Receptors According to Their Function in the Brain. Cell 2007, 131, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E.; Bhakoo, K.; Peuchen, S.; Dolphin, C.T.; Patel, R.; Clark, J.B. Distribution of mRNAs Encoding the Peroxisome Proliferator-Activated Receptor α, β, and γ and the Retinoid X Receptor α, β, and γ in Rat Central Nervous System. J. Neurochem. 1998, 70, 1366–1375. [Google Scholar] [CrossRef]
- Roy, A.; Jana, M.; Corbett, G.T.; Ramaswamy, S.; Kordower, J.H.; Gonzalez, F.J.; Pahan, K. Regulation of Cyclic AMP Response Element Binding and Hippocampal Plasticity-Related Genes by Peroxisome Proliferator-Activated Receptor α. Cell Rep. 2013, 4, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of Action of Fibrates on Lipid and Lipoprotein Metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- and, J.K.R.; Hashimoto, T. PEROXISOMAL β-OXIDATION AND PEROXISOME PROLIFERATOR–ACTIVATED RECEPTOR α: An Adaptive Metabolic System. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Schreurs, M.; Kuipers, F.; Van Der Leij, F.R. Regulatory enzymes of mitochondrial β-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev. 2010, 11, 380–388. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARα and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef] [Green Version]
- Semple, R.K.; Chatterjee, V.K.K.; O’Rahilly, S. PPARγ and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barish, G.D.; Narkar, V.A.; Evans, R.M. PPARδ: A dagger in the heart of the metabolic syndrome. J. Clin. Investig. 2006, 116, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvergne, B. PPARs special issue: Anchoring the present to explore the future. Biochim. Biophys. Acta (Bba) Mol. Cell Biol. Lipids 2007, 1771, 913–914. [Google Scholar] [CrossRef]
- Roy, A.; Kundu, M.; Jana, M.; Mishra, R.K.; Yung, Y.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. Identification and characterization of PPARα ligands in the hippocampus. Nat. Chem. Biol. 2016, 12, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef] [Green Version]
- Burris, T.P. PPARα ligands make memories. Nat. Chem. Biol. 2016, 12, 993–994. [Google Scholar] [CrossRef]
- Heneka, M.T.; Reyes-Irisarri, E.; Hüll, M.; Kummer, M.P. Impact and Therapeutic Potential of PPARs in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 643–650. [Google Scholar] [CrossRef]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Marco, F.; Francesca, F.; Maria Paola, C.; Sandra, M. Neuroprotective Properties of Peroxisome Proliferator-Activated Receptor Alpha (PPARα) and its Lipid Ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef]
- Aleshin, S.; Grabeklis, S.; Hanck, T.; Sergeeva, M.; Reiser, G. Peroxisome Proliferator-Activated Receptor (PPAR)-γ Positively Controls and PPARα Negatively Controls Cyclooxygenase-2 Expression in Rat Brain Astrocytes through a Convergence on PPARβ/δ via Mutual Control of PPAR Expression Levels. Mol. Pharmacol. 2009, 76, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Leisewitz, A.V.; Urrutia, C.R.; Martinez, G.R.; Loyola, G.; Bronfman, M. A PPARs cross-talk concertedly commits C6 glioma cells to oligodendrocytes and induces enzymes involved in myelin synthesis. J. Cell. Physiol. 2008, 217, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Reiser, G. Role of the peroxisome proliferator-activated receptors (PPAR)-α, β/δ and γ triad in regulation of reactive oxygen species signaling in brain. Biol. Chem. 2013, 394, 1553. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Strokin, M.; Sergeeva, M.; Reiser, G. Peroxisome proliferator-activated receptor (PPAR)β/δ, a possible nexus of PPARα- and PPARγ-dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem. Int. 2013, 63, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Glatz, T.; Stöck, I.; Nguyen-Ngoc, M.; Gohlke, P.; Herdegen, T.; Culman, J.; Zhao, Y. Peroxisome-proliferator-activated receptors γ and peroxisome-proliferator-activated receptors β/δ and the regulation of interleukin 1 receptor antagonist expression by pioglitazone in ischaemic brain. J. Hypertens. 2010, 28, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1, 217–233. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.-X.; Liu, F.; Iqbal, K. O-GlcNAcylation: A regulator of tau pathology and neurodegeneration. Alzheimer’s Dement. 2016, 12, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta (Bba)-Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer’s disease. J. Alzheimer’s Dis. 2005, 7, 45–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. The Link Between Tau and Insulin Signaling: Implications for Alzheimer’s Disease and Other Tauopathies. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Skerrett, R.; Pellegrino, M.P.; Casali, B.T.; Taraboanta, L.; Landreth, G.E. Combined Liver X Receptor/Peroxisome Proliferator-activated Receptor γ Agonist Treatment Reduces Amyloid β Levels and Improves Behavior in Amyloid Precursor Protein/Presenilin 1 Mice. J. Biol. Chem. 2015, 290, 21591–21602. [Google Scholar] [CrossRef] [Green Version]
- Escribano, L.; Simón, A.-M.; Gimeno, E.; Cuadrado-Tejedor, M.; López de Maturana, R.; García-Osta, A.; Ricobaraza, A.; Pérez-Mediavilla, A.; Del Río, J.; Frechilla, D. Rosiglitazone Rescues Memory Impairment in Alzheimer’s Transgenic Mice: Mechanisms Involving a Reduced Amyloid and Tau Pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Gold, M.; Gold, M.; Alderton, C.; Zvartau-Hind, M.; Egginton, S.; Saunders, A.M.; Irizarry, M.; Craft, S.; Landreth, G.; Linnamägi, Ü.; et al. Rosiglitazone Monotherapy in Mild-to-Moderate Alzheimer’s Disease: Results from a Randomized, Double-Blind, Placebo-Controlled Phase III Study. Dement. Geriatr. Cogn. Disord. 2010, 30, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef]
- Heneka, M.T.; Fink, A.; Doblhammer, G. Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 2015, 78, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.W.; Tanen, M.; Figueroa, D.J.; Biswas, C.; Zycband, E.; Moller, D.E.; Austin, C.P.; Berger, J.P. Localization of PPARδ in murine central nervous system: Expression in oligodendrocytes and neurons. Brain Res. 2003, 975, 10–21. [Google Scholar] [CrossRef]
- Sergey, K.; Jill, C.R.; Douglas, L.F. A PPARdelta Agonist Reduces Amyloid Burden and Brain Inflammation in a Transgenic Mouse Model of Alzheimers Disease. Curr. Alzheimer Res. 2009, 6, 431–437. [Google Scholar] [CrossRef]
- Tong, M.; Deochand, C.; Didsbury, J.; de la Monte, S.M. T3D-959: A Multi-Faceted Disease Remedial Drug Candidate for the Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 51, 123–138. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.; Gallucci, G.; Tong, M.; de la Monte, S.M. Therapeutic Advantages of Dual Targeting of PPAR-δ and PPAR-γ in an Experimental Model of Sporadic Alzheimer’s Disease. J. Parkinson’s Dis. Alzheimer’s Dis. 2018, 5. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Schiano, I.; Didsbury, J. Improved Brain Insulin/IGF Signaling and Reduced Neuroinflammation with T3D-959 in an Experimental Model of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, S.; Gabriel, H.; Strittmatter, W.; Didsbury, J. An Exploratory Phase IIa Study of the PPAR delta/gamma Agonist T3D-959 Assessing Metabolic and Cognitive Function in Subjects with Mild to Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1085–1103. [Google Scholar] [CrossRef] [Green Version]
- Murillo-Rodríguez, E.; Guzmán, K.; Arankowsky-Sandoval, G.; Salas-Crisóstomo, M.; Jiménez-Moreno, R.; Arias-Carrión, O. Evidence that activation of nuclear peroxisome proliferator-activated receptor alpha (PPARα) modulates sleep homeostasis in rats. Brain Res. Bull. 2016, 127, 156–163. [Google Scholar] [CrossRef]
- Mijangos-Moreno, S.; Poot-Aké, A.; Guzmán, K.; Arankowsky-Sandoval, G.; Arias-Carrión, O.; Zaldívar-Rae, J.; Sarro-Ramírez, A.; Murillo-Rodríguez, E. Sleep and neurochemical modulation by the nuclear peroxisome proliferator-activated receptor α (PPAR-α) in rat. Neurosci. Res. 2016, 105, 65–69. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, Y.-J.; Wang, H.; Song, L.; Huang, C.; Zhu, Q.; Wu, F.; Zhang, W. Antidepressant-like effects of fenofibrate in mice via the hippocampal brain-derived neurotrophic factor signalling pathway. Br. J. Pharmacol. 2017, 174, 177–194. [Google Scholar] [CrossRef]
- Li, M.; Wang, D.; Bi, W.; Jiang, Z.-e.; Piao, R.; Yu, H. Palmitoylethanolamide Exerts Antidepressant-Like Effects in Rats: Involvement of PPAR Pathway in the Hippocampus. J. Pharmacol. Exp. Ther. 2019, 369, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheggi, S.; Melis, M.; De Felice, M.; Aroni, S.; Muntoni, A.L.; Pelliccia, T.; Gambarana, C.; De Montis, M.G.; Pistis, M. PPARα modulation of mesolimbic dopamine transmission rescues depression-related behaviors. Neuropharmacology 2016, 110, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Huang, C.; Zhu, Q.; Tong, L.-J.; Zhang, W. WY14643 produces anti-depressant-like effects in mice via the BDNF signaling pathway. Psychopharmacology 2015, 232, 1629–1642. [Google Scholar] [CrossRef] [PubMed]
- Puligheddu, M.; Pillolla, G.; Melis, M.; Lecca, S.; Marrosu, F.; De Montis, M.G.; Scheggi, S.; Carta, G.; Murru, E.; Aroni, S.; et al. PPAR-Alpha Agonists as Novel Antiepileptic Drugs: Preclinical Findings. PLoS ONE 2013, 8, e64541. [Google Scholar] [CrossRef] [PubMed]
- Citraro, R.; Russo, E.; Leo, A.; Russo, R.; Avagliano, C.; Navarra, M.; Calignano, A.; De Sarro, G. Pharmacokinetic-pharmacodynamic influence of N-palmitoylethanolamine, arachidonyl-2′-chloroethylamide and WIN 55,212-2 on the anticonvulsant activity of antiepileptic drugs against audiogenic seizures in DBA/2 mice. Eur. J. Pharmacol. 2016, 791, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, N.; Vallée, L.; Lecointe, C.; Bouchaert, E.; Staels, B.; Bordet, R.; Auvin, S. Fenofibrate, a peroxisome proliferator–activated receptor-α agonist, exerts anticonvulsive properties. Epilepsia 2009, 50, 943–948. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Ma, A.; Zhang, X.; Li, W.; Yang, W.; Chen, C.; Jin, X. Orally administered oleoylethanolamide protects mice from focal cerebral ischemic injury by activating peroxisome proliferator-activated receptor α. Neuropharmacology 2012, 63, 242–249. [Google Scholar] [CrossRef]
- Sun, Y.; Alexander, S.P.H.; Garle, M.J.; Gibson, C.L.; Hewitt, K.; Murphy, S.P.; Kendall, D.A.; Bennett, A.J. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br. J. Pharmacol. 2007, 152, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Dotson, A.L.; Wang, J.; Chen, Y.; Manning, D.; Nguyen, H.; Saugstad, J.A.; Offner, H. Sex differences and the role of PPAR alpha in experimental stroke. Metab. Brain Dis. 2016, 31, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Dotson, A.L.; Wang, J.; Liang, J.; Nguyen, H.; Manning, D.; Saugstad, J.A.; Offner, H. Loss of PPARα perpetuates sex differences in stroke reflected by peripheral immune mechanisms. Metab. Brain Dis. 2016, 31, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.; Squassina, A.; Congiu, D.; Chillotti, C.; Niola, P.; Galderisi, S.; Pistis, M.; Del Zompo, M. Investigation of endocannabinoid system genes suggests association between peroxisome proliferator activator receptor-α gene (PPARA) and schizophrenia. Eur. Neuropsychopharmacol. 2013, 23, 749–759. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; La Rana, G.; Russo, R.; Sasso, O.; Iacono, A.; Esposito, E.; Raso, G.M.; Cuzzocrea, S.; Lo Verme, J.; Piomelli, D.; et al. Acute Intracerebroventricular Administration of Palmitoylethanolamide, an Endogenous Peroxisome Proliferator-Activated Receptor-α Agonist, Modulates Carrageenan-Induced Paw Edema in Mice. J. Pharmacol. Exp. Ther. 2007, 322, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuzzocrea, S.; Mazzon, E.; Di Paola, R.; Peli, A.; Bonato, A.; Britti, D.; Genovese, T.; Muià, C.; Crisafulli, C.; Caputi, A.P. The role of the peroxisome proliferator-activated receptor-α (PPAR-α) in the regulation of acute inflammation. J. Leukoc. Biol. 2006, 79, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orio, B.; Fracassi, A.; Ceru, M.P.; Moreno, S. Targeting PPARalpha in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 345–354. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Courchesne-Loyer, A.; Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Hennebelle, M.; Croteau, E.; Bocti, C.; Fulop, T.; Castellano, C.-A. Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-β-Hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Chen, S.; Mao, Z.; Cadenas, E.; Brinton, R.D. 2-Deoxy-D-Glucose Treatment Induces Ketogenesis, Sustains Mitochondrial Function, and Reduces Pathology in Female Mouse Model of Alzheimer’s Disease. PLoS ONE 2011, 6, e21788. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, C.; Medalie, J.; Scherma, M.; Panlilio, L.V.; Solinas, M.; Tanda, G.; Drago, F.; Cadet, J.L.; Goldberg, S.R.; Yasar, S. Fatty acid amide hydrolase (FAAH) inhibition enhances memory acquisition through activation of PPAR-alpha nuclear receptors. Learn Mem 2009, 16, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; You, Z.; Wu, Z.; Zhou, L.; Shen, J.; Gu, Z. WY14643 Attenuates the Scopolamine-Induced Memory Impairments in Mice. Neurochem. Res. 2016, 41, 2868–2879. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Cristiano, C.; Lyons, D.J.; Citraro, R.; Russo, E.; Avagliano, C.; Russo, R.; Raso, G.M.; Meli, R.; De Sarro, G.; et al. Peroxisome proliferator-activated receptor alpha plays a crucial role in behavioral repetition and cognitive flexibility in mice. Mol. Metab. 2015, 4, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Mounier, A.; Georgiev, D.; Nam, K.N.; Fitz, N.F.; Castranio, E.L.; Wolfe, C.M.; Cronican, A.A.; Schug, J.; Lefterov, I.; Koldamova, R. Bexarotene-Activated Retinoid X Receptors Regulate Neuronal Differentiation and Dendritic Complexity. J. Neurosci. 2015, 35, 11862–11876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.N.; Mounier, A.; Fitz, N.F.; Wolfe, C.; Schug, J.; Lefterov, I.; Koldamova, R. RXR controlled regulatory networks identified in mouse brain counteract deleterious effects of Aβ oligomers. Sci. Rep. 2016, 6, 24048. [Google Scholar] [CrossRef] [Green Version]
- Chikahisa, S.; Chida, D.; Shiuchi, T.; Harada, S.; Shimizu, N.; Otsuka, A.; Tanioka, D.; Séi, H. Enhancement of fear learning in PPARα knockout mice. Behav. Brain Res. 2019, 359, 664–670. [Google Scholar] [CrossRef]
- Brune, S.; Kölsch, H.; Ptok, U.; Majores, M.; Schulz, A.; Schlosser, R.; Rao, M.L.; Maier, W.; Heun, R. Polymorphism in the peroxisome proliferator-activated receptor α gene influences the risk for Alzheimer’s disease. J. Neural Transm. 2003, 110, 1041–1050. [Google Scholar] [CrossRef]
- Kölsch, H.; Lehmann, D.J.; Ibrahim-Verbaas, C.A.; Combarros, O.; van Duijn, C.M.; Hammond, N.; Belbin, O.; Cortina-Borja, M.; Lehmann, M.G.; Aulchenko, Y.S.; et al. Interaction of insulin and PPAR-α genes in Alzheimer’s disease: The Epistasis Project. J. Neural Transm. 2012, 119, 473–479. [Google Scholar] [CrossRef]
- Heun, R.; Kölsch, H.; Ibrahim-Verbaas, C.A.; Combarros, O.; Aulchenko, Y.S.; Breteler, M.; Schuur, M.; van Duijn, C.M.; Hammond, N.; Belbin, O.; et al. Interactions between PPAR-α and inflammation-related cytokine genes on the development of Alzheimer’s disease, observed by the Epistasis Project. Int. J. Mol. Epidemiol. Genet. 2012, 3, 39–47. [Google Scholar] [PubMed]
- Sjölander, A.; Minthon, L.; Bogdanovic, N.; Wallin, A.; Zetterberg, H.; Blennow, K. The PPAR-α gene in Alzheimer’s disease: Lack of replication of earlier association. Neurobiol. Aging 2009, 30, 666–668. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Maes, M.; Zambon, A. Fibrates and future PPARα agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 542–553. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Brown, W.V.; Dujovne, C.A.; Farquhar, J.W.; Feldman, E.B.; Grundy, S.M.; Knopp, R.H.; Lasser, N.L.; Mellies, M.J.; Palmer, R.H.; Samuel, P. Effects of fenofibrate on plasma lipids. Double-blind, multicenter study in patients with type IIA or IIB hyperlipidemia. Arterioscler.: Off. J. Am. Heart Assoc. Inc. 1986, 6, 670–678. [Google Scholar] [CrossRef]
- Brown, W.V. Focus on Fenofibrate. Hosp. Pract. 1988, 23, 31–40. [Google Scholar] [CrossRef]
- Hennuyer, N.; Duplan, I.; Paquet, C.; Vanhoutte, J.; Woitrain, E.; Touche, V.; Colin, S.; Vallez, E.; Lestavel, S.; Lefebvre, P.; et al. The novel selective PPARα modulator (SPPARMα) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016, 249, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor α agonists. Bioorg. Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.-C. Selective peroxisome proliferator-activated receptorα modulators (SPPARMα): The next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of Pemafibrate, a Novel Selective PPARα Modulator, on Lipid and Glucose Metabolism in Patients With Type 2 Diabetes and Hypertriglyceridemia: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, S.; Roy, A.; Jana, M.; Pahan, K. Cinnamic acid activates PPARα to stimulate Lysosomal biogenesis and lower Amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol. Dis. 2019, 124, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Su, L.-Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.-X.; Zhang, D.-F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Jana, M.; Kundu, M.; Corbett, G.T.; Rangaswamy, S.B.; Mishra, R.K.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. HMG-CoA Reductase Inhibitors Bind to PPARα to Upregulate Neurotrophin Expression in the Brain and Improve Memory in Mice. Cell Metab. 2015, 22, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Roy, A.; Kundu, M.; Jana, M.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proc. Natl. Acad. Sci. USA 2018, 115, E7408–E7417. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Jana, M.; Pahan, K. Aspirin Induces Lysosomal Biogenesis and Attenuates Amyloid Plaque Pathology in a Mouse Model of Alzheimer’s Disease via PPARα. J. Neurosci. 2018, 38, 6682–6699. [Google Scholar] [CrossRef]
- Medicine, N.L.O. Study Evaluating Potential Interaction Between SAM-531 And Gemfibrozil When Co-Administered. Available online: https://clinicaltrials.gov/ct2/show/NCT00966966 (accessed on 10 April 2020).
- Medicine, N.L.O. Modulation of Micro-RNA Pathways by Gemfibrozil in Predementia Alzheimer Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT02045056 (accessed on 10 April 2020).
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Wang, W.-X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The Expression of MicroRNA miR-107 Decreases Early in Alzheimer’s Disease and May Accelerate Disease Progression through Regulation of β-Site Amyloid Precursor Protein-Cleaving Enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.-X. MiR-107 is Reduced in Alzheimer’s Disease Brain Neocortex: Validation Study. J. Alzheimer’s Dis. 2010, 21, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haniu, M.; Denis, P.; Young, Y.; Mendiaz, E.A.; Fuller, J.; Hui, J.O.; Bennett, B.D.; Kahn, S.; Ross, S.; Burgess, T.; et al. Characterization of Alzheimer’s β-Secretase Protein BACE: A PEPSIN FAMILY MEMBER WITH UNUSUAL PROPERTIES. J. Biol. Chem. 2000, 275, 21099–21106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.-Z.; Sun, S.; Tan, C.-C.; Yu, J.-T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Willson, T.M.; Klockgether, T.; van Leuven, F.; Heneka, M.T. Nonsteroidal Anti-Inflammatory Drugs and Peroxisome Proliferator-Activated Receptor-γ Agonists Modulate Immunostimulated Processing of Amyloid Precursor Protein through Regulation of β-Secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar] [CrossRef] [Green Version]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.Z.; Usher, M.G.; Foley, E.L.; Milstone, D.S.; Brosius, F.C.; Mortensen, R.M. Sex dimorphic actions of rosiglitazone in generalised peroxisome proliferator-activated receptor-γ (PPAR-γ)-deficient mice. Diabetologia 2010, 53, 1493–1505. [Google Scholar] [CrossRef] [Green Version]
- Jalouli, M.; Carlsson, L.; Améen, C.; Lindén, D.; Ljungberg, A.; Michalik, L.; Edén, S.; Wahli, W.; Oscarsson, J. Sex Difference in Hepatic Peroxisome Proliferator-Activated Receptor α Expression: Influence of Pituitary and Gonadal Hormones. Endocrinology 2003, 144, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.E.; Ousman, S.S.; Sobel, R.A.; Zuniga, L.; Baranzini, S.E.; Youssef, S.; Crowell, A.; Loh, J.; Oksenberg, J.; Steinman, L. Peroxisome proliferator–activated receptor (PPAR)α expression in T cells mediates gender differences in development of T cell–mediated autoimmunity. J. Exp. Med. 2007, 204, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Maren, S.; De Oca, B.; Fanselow, M.S. Sex differences in hippocampal long-term potentiation (LTP) and Pavlovian fear conditioning in rats: Positive correlation between LTP and contextual learning. Brain Res. 1994, 661, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Zhang, K.; Xu, T.; Yamaki, V.N.; Wei, Z.; Huang, M.; Rose, G.M.; Cai, X. Sex Differences in Long-Term Potentiation at Temporoammonic-CA1 Synapses: Potential Implications for Memory Consolidation. PLoS ONE 2016, 11, e0165891. [Google Scholar] [CrossRef] [Green Version]
- Monfort, P.; Gomez-Gimenez, B.; Llansola, M.; Felipo, V. Gender Differences in Spatial Learning, Synaptic Activity, and Long-Term Potentiation in the Hippocampus in Rats: Molecular Mechanisms. ACS Chem. Neurosci. 2015, 6, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-W.; Pan, B.; Han, T.-Z.; Xie, W. Sexual dimorphism in the induction of LTP: Critical role of tetanizing stimulation. Life Sci. 2004, 75, 119–127. [Google Scholar] [CrossRef]
- Ris, L.; Angelo, M.; Plattner, F.; Capron, B.; Errington, M.L.; Bliss, T.V.P.; Godaux, E.; Giese, K.P. Sexual dimorphisms in the effect of low-level p25 expression on synaptic plasticity and memory. Eur. J. Neurosci. 2005, 21, 3023–3033. [Google Scholar] [CrossRef]
- Harte-Hargrove, L.C.; Varga-Wesson, A.; Duffy, A.M.; Milner, T.A.; Scharfman, H.E. Opioid Receptor-Dependent Sex Differences in Synaptic Plasticity in the Hippocampal Mossy Fiber Pathway of the Adult Rat. J. Neurosci. 2015, 35, 1723–1738. [Google Scholar] [CrossRef] [Green Version]
- Córdoba Montoya, D.A.; Carrer, H.F. Estrogen facilitates induction of long term potentiation in the hippocampus of awake rats1First published on the World Wide Web on 4 November 1997.1. Brain Res. 1997, 778, 430–438. [Google Scholar] [CrossRef]
- Good, M.; Day, M.; Muir, J.L. Cyclical changes in endogenous levels of oestrogen modulate the induction of LTD and LTP in the hippocampal CA1 region. Eur. J. Neurosci. 1999, 11, 4476–4480. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.M.; Lee, C.D. Estrogen replacement in ovariectomized rats affects strategy selection in the Morris water maze. Neurobiol. Learn. Mem. 2004, 82, 142–149. [Google Scholar] [CrossRef]
- Korol, D.L.; Malin, E.L.; Borden, K.A.; Busby, R.A.; Couper-Leo, J. Shifts in preferred learning strategy across the estrous cycle in female rats. Horm. Behav. 2004, 45, 330–338. [Google Scholar] [CrossRef]
- Gupta, R.R.; Sen, S.; Diepenhorst, L.L.; Rudick, C.N.; Maren, S. Estrogen modulates sexually dimorphic contextual fear conditioning and hippocampal long-term potentiation (LTP) in rats11Published on the World Wide Web on 1 December 2000. Brain Res. 2001, 888, 356–365. [Google Scholar] [CrossRef]
- Joffe, H.; Hall, J.E.; Gruber, S.; Sarmiento, I.A.; Cohen, L.S.; Yurgelun-Todd, D.; Martin, K.A. Estrogen therapy selectively enhances prefrontal cognitive processes: A randomized, double-blind, placebo-controlled study with functional magnetic resonance imaging in perimenopausal and recently postmenopausal women. Menopause 2006, 13, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.; Zhang, Q.-g.; Wang, R.; Vadlamudi, R.; Brann, D. Estrogen neuroprotection and the critical period hypothesis. Front. Neuroendocrinol. 2012, 33, 85–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Estrogen and Alzheimer’s disease: Still an attractive topic despite disappointment from early clinical results. Eur. J. Pharmacol. 2017, 817, 51–58. [Google Scholar] [CrossRef]
- Maher, A.C.; Akhtar, M.; Vockley, J.; Tarnopolsky, M.A. Women Have Higher Protein Content of β-Oxidation Enzymes in Skeletal Muscle than Men. PLoS ONE 2010, 5, e12025. [Google Scholar] [CrossRef] [Green Version]
- Wege, N.; Schutkowski, A.; Boenn, M.; Bialek, J.; Schlitt, A.; Noack, F.; Grosse, I.; Stangl, G.I. Men and women differ in their diurnal expression of monocyte peroxisome proliferator-activated receptor-α in the fed but not in the fasted state. FASEB J. 2015, 29, 2905–2911. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.A.; Rego, D.; Moshkova, M.; Kebir, H.; Chruscinski, A.; Nguyen, H.; Akkermann, R.; Stanczyk, F.Z.; Prat, A.; Steinman, L.; et al. Peroxisome proliferator-activated receptor (PPAR)α and -γ regulate IFNγ and IL-17A production by human T cells in a sex-specific way. Proc. Natl. Acad. Sci. USA 2012, 109, 9505–9510. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Appelman, Y.; van Rijn, B.B.; ten Haaf, M.E.; Boersma, E.; Peters, S.A.E. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis 2015, 241, 211–218. [Google Scholar] [CrossRef]
- Brinton, R.D. Estrogen-induced plasticity from cells to circuits: Predictions for cognitive function. Trends Pharmacol. Sci. 2009, 30, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Day, M.; Muñiz, L.C.; Bitran, D.; Arias, R.; Revilla-Sanchez, R.; Grauer, S.; Zhang, G.; Kelley, C.; Pulito, V.; et al. Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 2008, 11, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R.S.; Diaz Brinton, R.; Mosconi, L. Female Sex and Alzheimer’s Risk: The Menopause Connection. J. Prev. Alzheimer’s Dis. 2018, 5, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Jackson, H.; Hristov, H.; Isaacson, R.S.; Saif, N.; Shetty, T.; Etingin, O.; Henchcliffe, C.; Brinton, R.D.; Mosconi, L. Sex and Gender Driven Modifiers of Alzheimer’s: The Role for Estrogenic Control Across Age, Race, Medical, and Lifestyle Risks. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Akama, K.T.; Spencer-Segal, J.L.; Milner, T.A.; Waters, E.M. Estrogen effects on the brain: Actions beyond the hypothalamus via novel mechanisms. Behav. Neurosci. 2012, 126, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Brinton, R.D. Estrogen Regulation of Mitochondrial Bioenergetics: Implications for Prevention of Alzheimer’s Disease. In Advances in Pharmacolog; Michaelis, E.K., Michaelis, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 64, pp. 327–371. [Google Scholar]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Benz, V.; Kintscher, U.; Foryst-Ludwig, A. Sex-Specific Differences in Type 2 Diabetes Mellitus and Dyslipidemia Therapy: PPAR Agonists. In Sex and Gender Differences in Pharmacology; Regitz-Zagrosek, V., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 387–410. [Google Scholar]
- Wong, M.W.K.; Braidy, N.; Crawford, J.; Pickford, R.; Song, F.; Mather, K.A.; Attia, J.; Brodaty, H.; Sachdev, P.; Poljak, A. APOE Genotype Differentially Modulates Plasma Lipids in Healthy Older Individuals, with Relevance to Brain Health. J. Alzheimer’s Dis. 2019, 72, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Fabelo, N.; Ferrer, I.; Marín, R. “Lipid raft aging” in the human frontal cortex during nonpathological aging: Gender influences and potential implications in Alzheimer’s disease. Neurobiol. Aging 2018, 67, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Haughey, N.J.; Han, D.; An, Y.; Bandaru, V.V.R.; Lyketsos, C.G.; Ferrucci, L.; Resnick, S.M. The Association Between Plasma Ceramides and Sphingomyelins and Risk of Alzheimer’s Disease Differs by Sex and APOE in the Baltimore Longitudinal Study of Aging. J. Alzheimer’s Dis. 2017, 60, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Couttas, T.A.; Kain, N.; Tran, C.; Chatterton, Z.; Kwok, J.B.; Don, A.S. Age-Dependent Changes to Sphingolipid Balance in the Human Hippocampus are Gender-Specific and May Sensitize to Neurodegeneration. J. Alzheimer’s Dis. 2018, 63, 503–514. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sáez-Orellana, F.; Octave, J.-N.; Pierrot, N. Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α. Cells 2020, 9, 1215. https://doi.org/10.3390/cells9051215
Sáez-Orellana F, Octave J-N, Pierrot N. Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α. Cells. 2020; 9(5):1215. https://doi.org/10.3390/cells9051215
Chicago/Turabian StyleSáez-Orellana, Francisco, Jean-Noël Octave, and Nathalie Pierrot. 2020. "Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α" Cells 9, no. 5: 1215. https://doi.org/10.3390/cells9051215
APA StyleSáez-Orellana, F., Octave, J. -N., & Pierrot, N. (2020). Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α. Cells, 9(5), 1215. https://doi.org/10.3390/cells9051215