Consequences of Lmna Exon 4 Mutations in Myoblast Function

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and 3D Model
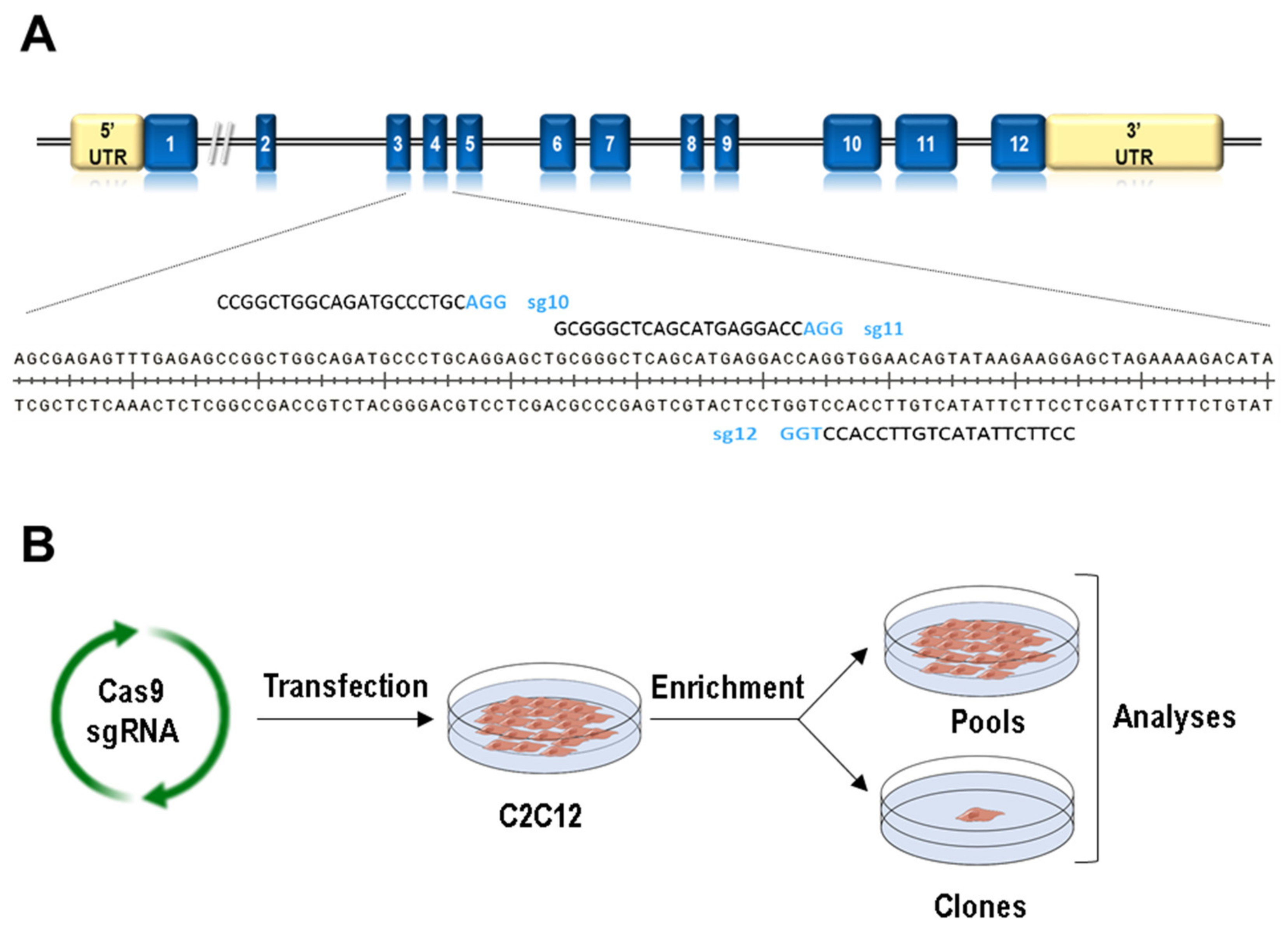
2.2. Generation of Clones with Mutations in Lmna Exon 4 Using CRISPR/Cas9
2.3. DNA Sequencing and Bioinformatic Analysis
2.4. Western Blot Analysis
2.5. Immunofluorescence Microscopy, Nuclear Morphology Analyses, and Myogenic Differentiation Studies
2.6. Quantification and Statistical Analysis
3. Results
3.1. Generation of Lmna Exon 4 Mutations in C2C12 Cells
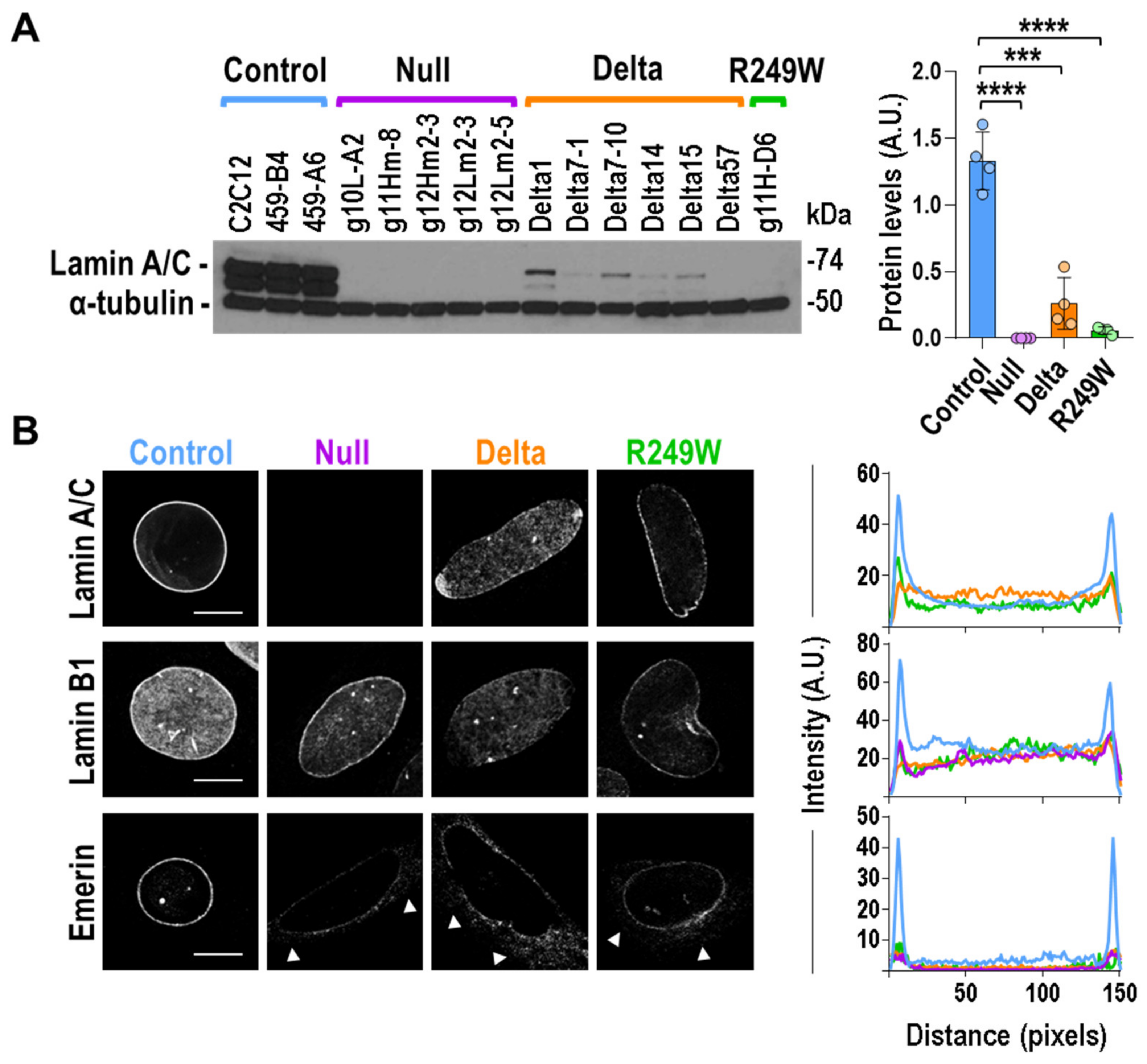
3.2. Components of the Nuclear Lamina Were Abnormally Distributed in Lmna Exon 4 Mutant Myoblasts, Lamin A/C Abnormal Expression, and Subcellular Localization in Lmna Exon 4 Mutants
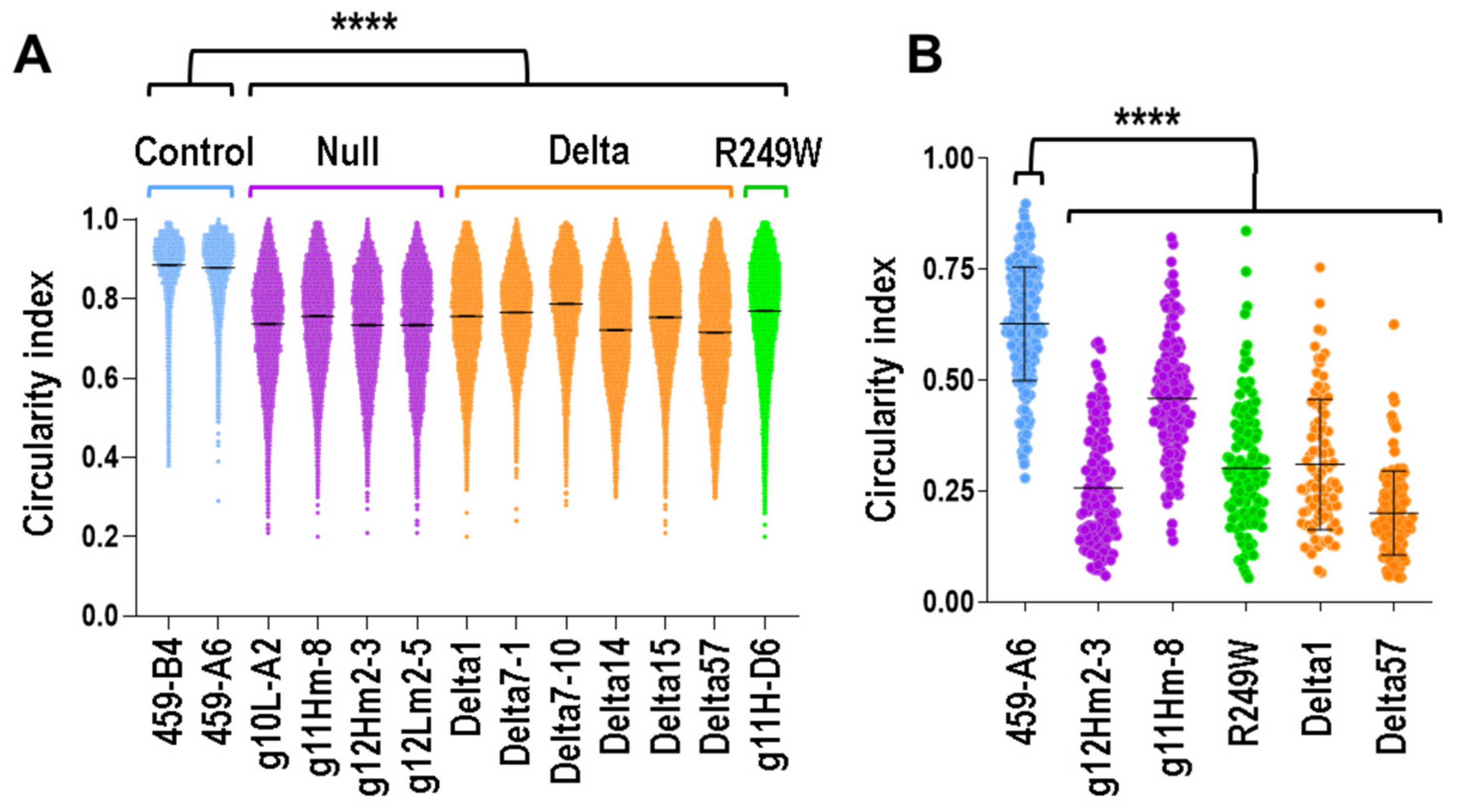
3.3. Nuclear Morphology Abnormalities Were Common for All the Clones with Mutations in Lmna Exon 4
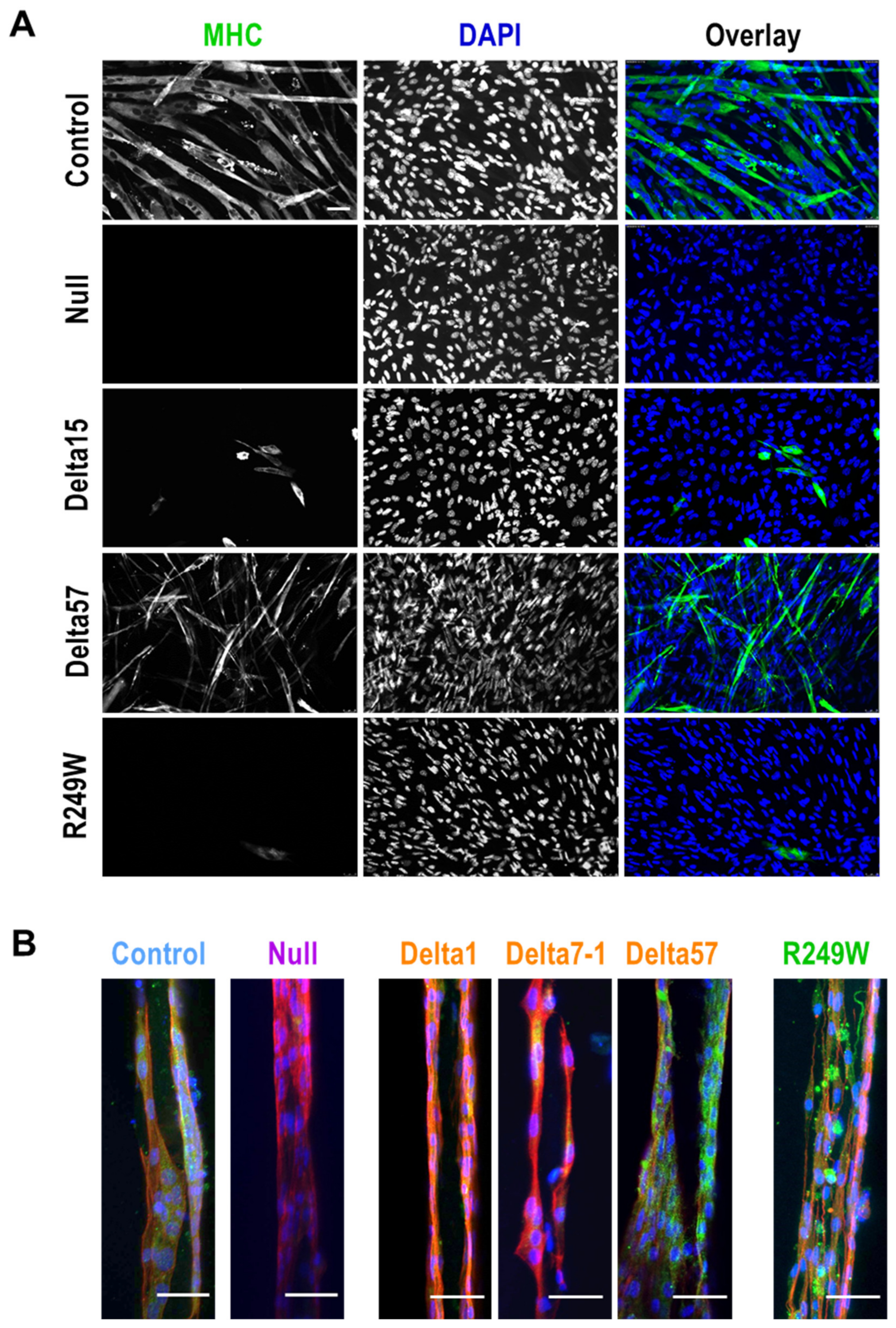
3.4. Abnormal Myogenic Differentiation Was Common to All Myoblasts Carrying Lmna Exon 4 Mutations
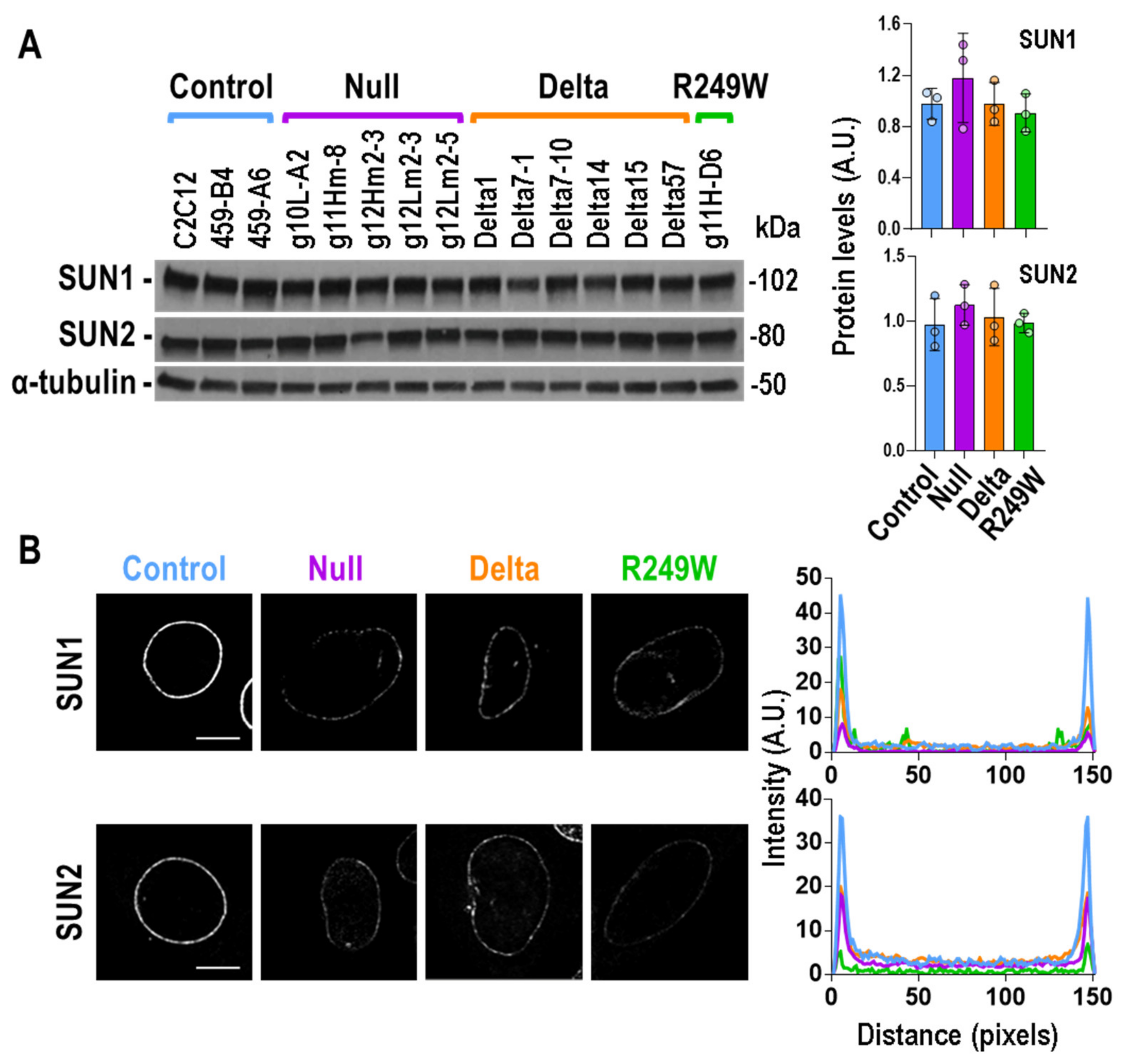
3.5. Expression of Lmna Exon 4 Mutants Was Associated with Abnormal Subcellular Localization of SUN1 and SUN2
3.6. MAPK Signaling Was Not Altered in Lmna Exon 4 Mutant Myoblasts
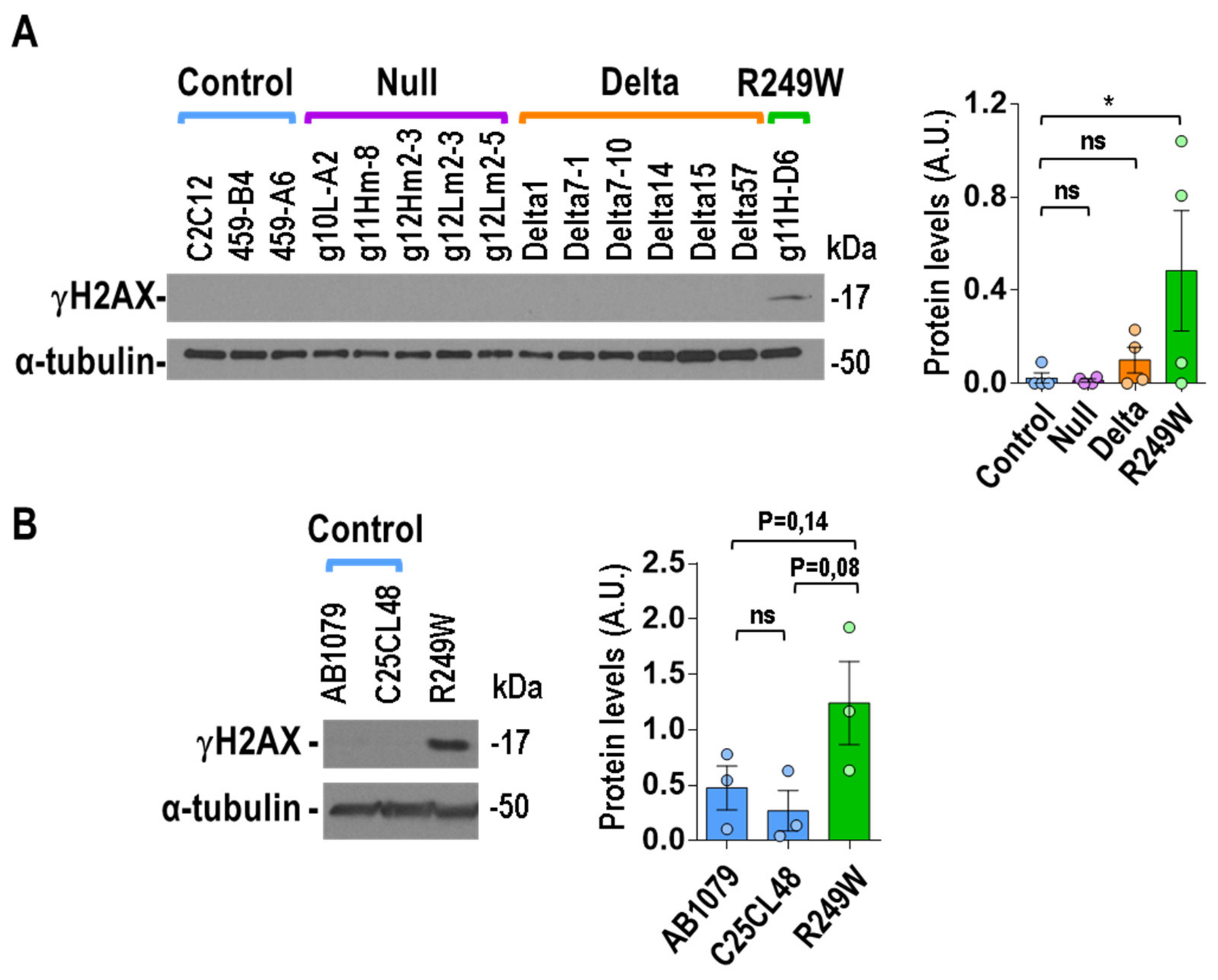
3.7. High Levels of DNA Damage Were Associated with R249W Mutation
4. Discussion
4.1. Comparison of Lmna Exon 4 Mutants Generated in This Study with Previously Reported LMNA Mutants
4.2. Mechanistic and Functional Defects Associated with Lmna Exon 4 Mutations
4.3. DNA Damage: A Feature Specific for the R249W Mutation?
4.4. CRISPR/Cas9 Activity and Gene Therapy Implications
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Synthesis of Prepolymer Precursors
Preparation of Prepolymer Solutions
References
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.L.; Zullo, J.M.; Bertolino, E.; Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008, 452, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, J.; Discher, D.E. The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J. Cell Sci. 2014, 127, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Busch, A.; Kiel, T.; Heupel, W.-M.; Wehnert, M.; Hübner, S. Nuclear protein import is reduced in cells expressing nuclear envelopathy-causing lamin A mutants. Exp. Cell Res. 2009, 315, 2373–2385. [Google Scholar] [CrossRef]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Cenni, V.; Capanni, C.; Mattioli, E.; Columbaro, M.; Wehnert, M.; Ortolani, M.; Fini, M.; Novelli, G.; Bertacchini, J.; Maraldi, N.M.; et al. Rapamycin treatment of Mandibuloacral dysplasia cells rescues localization of chromatin-associated proteins and cell cycle dynamics. Aging 2014, 6, 755–770. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Columbaro, M.; Capanni, C.; D’Apice, M.R.; Cavallo, C.; Murdocca, M.; Lattanzi, G.; Squarzoni, S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget 2015, 6, 29914–29928. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest. 2007, 117, 1282–1293. [Google Scholar] [CrossRef]
- Muchir, A.; Wu, W.; Choi, J.C.; Iwata, S.; Morrow, J.; Homma, S.; Worman, H.J. Abnormal p38 mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2012, 21, 4325–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical Inhibition of NAT10 Corrects Defects of Laminopathic Cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyret, E.; Liao, H.-K.; Yamamoto, M.; Hernandez-Benitez, R.; Fu, Y.; Erikson, G.; Reddy, P.; Izpisua Belmonte, J.C. Single-dose CRISPR–Cas9 therapy extends lifespan of mice with Hutchinson–Gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Fernández, O.; Osorio, F.G.; Quesada, V.; Rodríguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R.; et al. Development of a CRISPR/Cas9-based therapy for Hutchinson–Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Brull, A.; Azibani, F.; Benarroch, L.; Chikhaoui, K.; Stewart, C.L.; Medalia, O.; Ben Yaou, R.; Bonne, G. Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery-Dreifuss Muscular Dystrophy. Cells 2020, 9, 844. [Google Scholar] [CrossRef] [Green Version]
- Scharner, J.; Brown, C.A.; Bower, M.; Iannaccone, S.T.; Khatri, I.A.; Escolar, D.; Gordon, E.; Felice, K.; Crowe, C.A.; Grosmann, C.; et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum. Mutat. 2011, 32, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front. Physiol. 2018, 9, 1332. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2019, 19, 464–473. [Google Scholar] [CrossRef]
- Scharner, J.; Figeac, N.; Ellis, J.A.; Zammit, P.S. Ameliorating pathogenesis by removing an exon containing a missense mutation: A potential exon-skipping therapy for laminopathies. Gene Ther. 2015, 22, 503–515. [Google Scholar] [CrossRef]
- Yaffe, D.; Saxel, O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977, 270, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Fernández-Garibay, X.; Castaño, A.G.; De Chiara, F.; Hernández-Albors, A.; Balaguer-Trias, J.; Ramón-Azcón, J. Muscle-on-a-chip with an on-site multiplexed biosensing system for in situ monitoring of secreted IL-6 and TNF-α. Lab. Chip 2019, 19, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C.; Franch, M.; Tabas-Madrid, D.; San-León, D.; Montoliu, L.; Cubas, P.; Pazos, F. Breaking-Cas—interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 2016, 44, W267–W271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, A.; Alvarez-Reyes, M.; Bridger, J.M.; Broers, J.L.; Ramaekers, F.C.; Wehnert, M.; Morris, G.E.; Whitfield, W.G.F.; Hutchison, C.J. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 2001, 114, 2577–2590. [Google Scholar]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; van Engelen, B.G.; Lammens, M.; Mislow, J.M.; McNally, E.; Schwartz, K.; Bonne, G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 2003, 291, 352–362. [Google Scholar] [CrossRef]
- Hale, C.M.; Shrestha, A.L.; Khatau, S.B.; Stewart-Hutchinson, P.J.; Hernandez, L.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Dysfunctional Connections Between the Nucleus and the Actin and Microtubule Networks in Laminopathic Models. Biophys. J. 2008, 95, 5462–5475. [Google Scholar] [CrossRef] [Green Version]
- Favreau, C.; Higuet, D.; Courvalin, J.-C.; Buendia, B. Expression of a Mutant Lamin A That Causes Emery-Dreifuss Muscular Dystrophy Inhibits In Vitro Differentiation of C2C12 Myoblasts. Mol. Cell. Biol. 2004, 24, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, E.; Ledran, M.; Hutchison, C.J. Remodelling of the nuclear lamina and nucleoskeleton is required for skeletal muscle differentiation in vitro. J. Cell Sci. 2005, 118, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Meinke, P.; Thuy, D.N.; Wehnert, M.S. The LINC complex and human disease. Biochem. Soc. Trans. 2011, 39, 1693–1697. [Google Scholar] [CrossRef] [Green Version]
- Gerbino, A.; Procino, G.; Svelto, M.; Carmosino, M. Role of Lamin A/C Gene Mutations in the signaling defects leading to cardiomyopathies. Front. Physiol. 2018, 9, 1356. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Wu, W.; Muchir, A.; Iwata, S.; Homma, S.; Worman, H.J. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem. 2012, 287, 40513–40524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burla, R.; La Torre, M.; Merigliano, C.; Vernì, F.; Saggio, I. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus 2018, 9, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziano, S.; Kreienkamp, R.; Coll-Bonfill, N.; Gonzalo, S. Causes and consequences of genomic instability in laminopathies: Replication stress and interferon response. Nucleus 2018, 9, 289–306. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.N.; Lombardi, R.; Karmouch, J.; Tsai, J.Y.; Czernuszewicz, G.; Taylor, M.R.G.; Mestroni, L.; Coarfa, C.; Gurha, P.; Marian, A.J. DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated with LMNA (Lamin A/C) Mutations. Circ. Res. 2019, 124, 856–873. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Chiu, P.H.; Yip, K.Y.; Chan, S.Y. Subcellular Localization of SUN2 Is Regulated by Lamin A and Rab5. PLoS ONE 2011, 6, e20507. [Google Scholar] [CrossRef] [Green Version]
- Méjat, A.; Decostre, V.; Li, J.; Renou, L.; Kesari, A.; Hantaï, D.; Stewart, C.L.; Xiao, X.; Hoffman, E.; Bonne, G.; et al. Lamin A/C–mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J. Cell Biol. 2009, 184, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 Interacts with Nuclear Lamin A and Cytoplasmic Nesprins To Provide a Physical Connection between the Nuclear Lamina and the Cytoskeleton. Mol. Cell. Biol. 2006, 26, 3738. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.; Güttinger, S.; Mühlhäusser, P.; Anderegg, F.; Bürgler, S.; Kutay, U. Nuclear envelope localization of human UNC84A does not require nuclear lamins. FEBS Lett. 2006, 580, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chi, Y.-H.; Mutalif, R.A.; Starost, M.F.; Myers, T.G.; Anderson, S.A.; Stewart, C.L.; Jeang, K.-T. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 2012, 149, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostlund, C.; Folker, E.S.; Choi, J.C.; Gomes, E.R.; Gundersen, G.G.; Worman, H.J. Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J. Cell Sci. 2009, 122, 4099–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Cenni, V.; Scotlandi, K.; Marino, M.T.; Merlini, L.; Squarzoni, S.; Lattanzi, G. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011, 18, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Wu, W.; Worman, H.J. Reduced expression of A-type lamins and emerin activates extracellular signal-regulated kinase in cultured cells. Biochim. Biophys. Acta 2009, 1792, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frock, R.L.; Kudlow, B.A.; Evans, A.M.; Jameson, S.A.; Hauschka, S.D.; Kennedy, B.K. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006, 20, 486–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janin, A.; Bauer, D.; Ratti, F.; Valla, C.; Bertrand, A.; Christin, E.; Chopin, E.; Streichenberger, N.; Bonne, G.; Gache, V.; et al. SMAD6 overexpression leads to accelerated myogenic differentiation of LMNA mutated cells. Sci. Rep. 2018, 8, 5618. [Google Scholar] [CrossRef]
- Liu, Q.; Pante, N.; Misteli, T.; Elsagga, M.; Crisp, M.; Hodzic, D.; Burke, B.; Roux, K.J. Functional association of Sun1 with nuclear pore complexes. J. Cell Biol. 2007, 178, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Jahed, Z.; Soheilypour, M.; Peyro, M.; Mofrad, M.R.K. The LINC and NPC relationship—It’s complicated! J. Cell Sci. 2016, 129, 3219–3229. [Google Scholar] [CrossRef] [Green Version]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 2017, 26, 333–343. [Google Scholar] [CrossRef]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Liang, H.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I.; Kasapi, M.; et al. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camozzi, D.; Capanni, C.; Cenni, V.; Mattioli, E.; Columbaro, M.; Squarzoni, S.; Lattanzi, G. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Nucleus 2014, 5, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Musich, P.R.; Zou, Y. DNA-damage accumulation and replicative arrest in Hutchinson–Gilford progeria syndrome. Biochem. Soc. Trans. 2011, 39, 1764–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, B.; Zitnik, G.; Johnson, S.; Nguyen, Q.; Risques, R.A.; Martin, G.M.; Oshima, J. DNA damage accumulation and TRF2 degradation in atypical Werner syndrome fibroblasts with LMNA mutations. Front. Genet. 2013, 4, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, C.M.; Wang, L.; Alcalai, R.; Pizard, A.; Burgon, P.G.; Ahmad, F.; Sherwood, M.; Branco, D.M.; Wakimoto, H.; Fishman, G.I.; et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell. Cardiol. 2008, 44, 293–303. [Google Scholar] [CrossRef] [Green Version]
- García-Lizarribar, A.; Fernández-Garibay, X.; Velasco-Mallorquí, F.; Castaño, A.G.; Samitier, J.; Ramon-Azcon, J. Composite Biomaterials as Long-Lasting Scaffolds for 3D Bioprinting of Highly Aligned Muscle Tissue. Macromol. Biosci. 2018, 18, 1–13. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ANTIBODIES | |||
|---|---|---|---|
| Name | Source (identifier) | Western Blot | Immuno- Fluorescence |
| Anti-mouse Lamin A/C (E-1) | Santa Cruz Biotechnology (Dallas, Texas, USA) (sc-376248) | 1:3000 | 1:500 |
| Anti-mouse α-tubulin | Sigma-Aldrich (St. Louis, MI, USA) (T9026) | 1:5000 | |
| Anti-rabbit lamin B1 | Abcam (Cambridge, UK) (ab16048) | 1:100 | |
| Anti-rabbit emerin (D3B9G) | Cell Signaling (Danvers, MA, USA) (#30853) | 1:100 | |
| Anti-mouse Sad1 And UNC84 Domain Containing 1 (SUN1) BBmSun1 IgC2b X12.11 | Donated by Dr. Colin Stewart | 1:50 | 1:50 |
| Anti-rabbit SUN2 (Sad1 And UNC84 Domain Containing 2) 11905 | Donated by Dr. Eric Schirmer | 1:500 | 1:200 |
| Anti-rabbit p44/42 Mitogen-Activated Protein Kinase 1/3 (Erk1/2)-137F5 | Cell Signaling (Danvers, MA, USA) (#4695) | 1:1000 | |
| Anti-rabbit phospho-p44/42 MAPK Erk1/2 Thr202/Tyr204 D13.14.4E | Cell Signaling (Danvers, MA, USA) (#4370) | 1:1000 | |
| Anti-mouse Akt (pan) 40D4 | Cell Signaling (Danvers, MA, USA) (#2920) | 1:1000 | |
| Anti-rabbit phospho-Akt (Ser473) D9E | Cell Signaling (Danvers, MA, USA) (#4060) | 1:1000 | |
| Anti-mouse phospho-histone H2A.X (Ser139) | Millipore (Burlington, MA, USA) (05-636-I) | 1:500 | |
| Anti-MYH7 (Myosin Heavy Chain 7) | Thermofisher (Waltham, MA, USA) (PA5-69132) | 1:200 | |
| MF20 (Mouse Monoclonal Anti-Myosin Heavy Chain Antibody) | DSHB Hybridoma (Iowa City, Iowa, USA) | 1:50 | |
| Rhodamine phalloidin | Thermofisher (Waltham, MA, USA) (R415) | 1:40 | |
| HRP-labelled anti-mouse secondary antibody | GE Healthcare (Chicago, Illinois, USA) (NA931-1ML) | 1:5000 | |
| HRP-labelled anti-rabbit secondary antibody | GE Healthcare (Chicago, Illinois, USA) (NA934-1ML) | 1:5000 | |
| Goat anti-mouse Alexa Fluor 488 | Thermofisher Scientific (Waltham, MA, USA) (A32723) | 1:500 | |
| Goat anti-rabbit Alexa Fluor 594 | Thermofisher Scientific (Waltham, MA, USA) (A32740) | 1:500 | |
| Goat anti-rabbit Alexa Fluor 488 | Thermofisher Scientific (Waltham, MA, USA) (A32731) | 1:500 | |
| Goat anti-rabbit Alexa Fluor 488 (3D models) | Thermofisher (Waltham, MA, USA) (A11034) | 1:200 | |
| CELL CULTURE MEDIA | REAGENTS and PLASMIDS | ||
| C2C12 myoblasts/SIGMA (St. Louis, MI, USA) (91031101) | pX459 vector (pSpCas9(BB)-2A-Puro)/Addgene (Watertown, MA, USA) (#62988) | ||
| DMEM (Dulbecco’s modified Eagle’s medium) high glucose/Invitrogen (Waltham, MA, USA) (61965-026) | Lipofectamine 3000/Invitrogen (Waltham, MA, USA) (L3000015) | ||
| FBS (fetal bovine serum)/Sigma-Aldrich (St. Louis, MI, USA) (#F7524-500ML) | Puromycin/InvivoGen (San Diego, CA, USA) (ant-pr-1) | ||
| Penicillin/streptomycin/Lonza (Basel, Switzerland) (#DE17-602E) | DNA polymerase/NZYTech (Lisbon, Portugal) (MB354) | ||
| Medium 199/Invitrogen (Waltham, MA, USA) (41150020) | MiSeq DNA/Illumina (San Diego, CA, USA) (MS-102-2003) | ||
| Fetuin/Life Technologies (Waltham, MA, USA) (10344026) | BCA system/Pierce (Waltham, MA, USA) (23227) | ||
| hEGF/Life Technologies (Waltham, MA, USA) (PHG0311) | ECL western blotting system/Thermo Fisher Scientific (Waltham, MA, USA) (Pierce 32106) | ||
| bFGF/Life Technologies (Waltham, MA, USA) (PHG0026) | Methanol/Panreac AppliChem (Barcelona, Spain) (#131091.1612) | ||
| Insuline/Sigma (St. Louis, MI, USA) (91077C-1G) | BSA (bovine serum albumin)/Sigma-Aldrich (St. Louis, MI, USA) (#A7906) | ||
| Dexamethasone/Sigma (St. Louis, MI, USA) (D4902-100mg) | PBS (phosphate-buffered saline)/Lonza (Basel, Switzerland) (#BE17-515Q) | ||
| Horse serum/Thermofisher (Waltham, MA, USA) (#26050-088) | Goat serum/Sigma-Aldrich (St. Louis, MI, USA) (#G9023-10ML) | ||
| SOFTWARE AND PLATFORMS | Donkey serum/Sigma-Aldrich (St. Louis, MI, USA) (#D9663-10ML) | ||
| CRISPResso (http://crispresso.pinellolab.partners.org/) | Triton X-100/Sigma-Aldrich (St. Louis, MI, USA) (#X100-1L) | ||
| TIDE (https://tide.deskgen.com/) | Prolong Gold with 4′,6-Diamidino-2-Phenylindole (DAPI)/Cell Signalling Technology (Danvers, MA, USA) (P36935) | ||
| ImageJ (U.S. National Institutes of Health, Bethesda, Maryland, USA) | TBS (Tris-buffered saline)/Canvax Biotech (Cordoba, Spain) (BR0042) | ||
| Prism 8 (GraphPad Software, Inc) | Hoechst 33324/Thermo Fisher Scientific (Waltham, MA, USA) (H3570) | ||
| Name and 5′ to 3′ Sequence |
|---|
| sg10: CCGGCTGGCAGATGCCCTGCAGG |
| sg11: GCGGGCTCAGCATGAGGACCAGG |
| sg12: GGTCCACCTTGTCATATTCTTCC |
| ssODNmex4g10: GTGGAGATCGATAACGGGAAGCAGCGAGAGTTTGAGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCTCAGCATGAGGACCAGGTGGAACAGTATAAGAAGGAGCT |
| ssODNmex4g11: GAGTTTGAGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCTCAGCATGAGGACCAGGTGGAACAGTATAAGAAGGAGCTAGAAAAGACATACTCCGCCAAGGTGCT |
| ssODNmex4g12: AGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCTCAGCATGAGGACCAGGTGGAACAGTATAAGAAGGAGCTAGAAAAGACATACTCCGCCAAGTGCTGGCCTCAT |
| ssODNmex4g10mut: GTGGAGATCGATAACGGGAAGCAGCGAGAGTTTGAGAGCCGGCTGGCAGATGCCCTGCAAGAGCTCTGGGCTCAGCATGAGGACCAGGTGGAACAGTATAAGAAGGAGCT |
| ssODNmex4g10mut2: GTGGAGATCGATAACGGGAAGCAGCGAGAGTTTGAGAGCCGTCTTGCCGACGCACTTCAAGAGCTCTGGGCTCAGCATGAGGACCAGGTGGAACAGTATAAGAAGGAGCT |
| ssODNmex4g11mut: GAGTTTGAGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCTCAGCATGAGGACCAAGTGGAACAGTATAAGAAGGAGCTAGAAAAGACATACTCCGCCAAGGTGCT |
| ssODNmex4g11mut2: GAGTTTGAGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCACAACACGAAGATCAAGTGGAACAGTATAAGAAGGAGCTAGAAAAGACATACTCCGCCAAGGTGCT |
| ssODNmex4g12mut: AGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCTCAGCATGAGGATCAGGTGGAACAGTATAAGAAGGAGCTAGAAAAGACATACTCCGCCAAGTGCTGGCCTCAT |
| ssODNmex4g12mut2: AGAGCCGGCTGGCAGATGCCCTGCAGGAGCTCTGGGCTCAGCATGAGGATCATGTTGAGCAATACAAAAAAGAGCTAGAAAAGACATACTCCGCCAAGTGCTGGCCTCAT |
| DeepSeq-Fw: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGGCGAGTGGATGCTGAG |
| DeepSeq-Rv: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGTCAATGCGGATTCGAGACT |
| Sanger-mLmna_Ex4_Fw: CCAGGCTAAGCGAGGGCTGC |
| Sanger-mLmna_Ex4_Rv: CCTGAGGAAGGCATCCCTGG |
| ID | Type | CDS (1) | Protein (Expected) |
|---|---|---|---|
| 459-A6 | Control | wt/wt | p.(666*)/p.(666*) (665 aa) |
| 459-B4 | wt/wt | p.(666*)/p.(666*) (665 aa) | |
| g10L-A2 | Null | c. 734_735del | p.(252*) (251 aa) |
| g11Hm-8 | c. [757_758ins] [758_810del] | p.(263*) (262 aa) | |
| g12Hm2-3 | c. [767del] [811subsC > G] | p.(263*) (262 aa) | |
| g12Lm2-3 | c. [739_740ins] [740_761del] [766_767GT > AG] | p.(273*) (272 aa) | |
| g12Lm2-5 | c. [del767]/c. [del769] | p.(263*) (262 aa)/p.(263*) (262 aa) | |
| g10L-A1 | Delta1 | c.734_736del (loss of 3 nt) | p.Leu245del (loss of 1 aa) |
| g11Hm2-1 | Delta7-1 | c.754_774del (loss of 21 nt) | p.His252_Gln258del (loss of 7 aa) |
| g11Hm2-10 | Delta7-10 | c.754_774del (loss of 21 nt) | p.His252_Gln258del (loss of 7 aa) |
| g10Hm-5 | Delta14 | c.707_748del (loss of 42 nt) | p.Glu236_Arg249del (loss of 14 aa) |
| g12Hm-6 | Delta15 | c.766_810del (loss of 45 nt) | p.Val256_Lys270del (loss of 15 aa) |
| g10H-A4 | Delta57 | c. [726_727ins] [727_1967del]/c. 640_810del | p.(252*) (251 aa)/p.Glu214_Lys270del (loss of 57 aa) |
| g11H-D6 | R249W | c. [744_745subsGC > CT] [750subsT > G] | p.Arg249Tryp (665 aa) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Domínguez, D.; Epifano, C.; de Miguel, F.; Castaño, A.G.; Vilaplana-Martí, B.; Martín, A.; Amarilla-Quintana, S.; Bertrand, A.T.; Bonne, G.; Ramón-Azcón, J.; et al. Consequences of Lmna Exon 4 Mutations in Myoblast Function. Cells 2020, 9, 1286. https://doi.org/10.3390/cells9051286
Gómez-Domínguez D, Epifano C, de Miguel F, Castaño AG, Vilaplana-Martí B, Martín A, Amarilla-Quintana S, Bertrand AT, Bonne G, Ramón-Azcón J, et al. Consequences of Lmna Exon 4 Mutations in Myoblast Function. Cells. 2020; 9(5):1286. https://doi.org/10.3390/cells9051286
Chicago/Turabian StyleGómez-Domínguez, Déborah, Carolina Epifano, Fernando de Miguel, Albert García Castaño, Borja Vilaplana-Martí, Alberto Martín, Sandra Amarilla-Quintana, Anne T Bertrand, Gisèle Bonne, Javier Ramón-Azcón, and et al. 2020. "Consequences of Lmna Exon 4 Mutations in Myoblast Function" Cells 9, no. 5: 1286. https://doi.org/10.3390/cells9051286
APA StyleGómez-Domínguez, D., Epifano, C., de Miguel, F., Castaño, A. G., Vilaplana-Martí, B., Martín, A., Amarilla-Quintana, S., Bertrand, A. T., Bonne, G., Ramón-Azcón, J., Rodríguez-Milla, M. A., & Pérez de Castro, I. (2020). Consequences of Lmna Exon 4 Mutations in Myoblast Function. Cells, 9(5), 1286. https://doi.org/10.3390/cells9051286