Axonal Transport as an In Vivo Biomarker for Retinal Neuropathy


 ,
,
Abstract
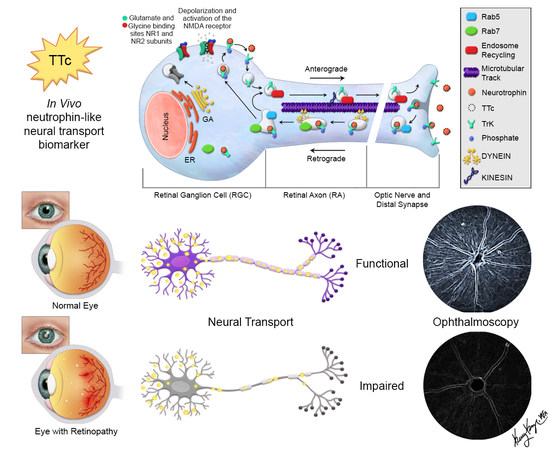
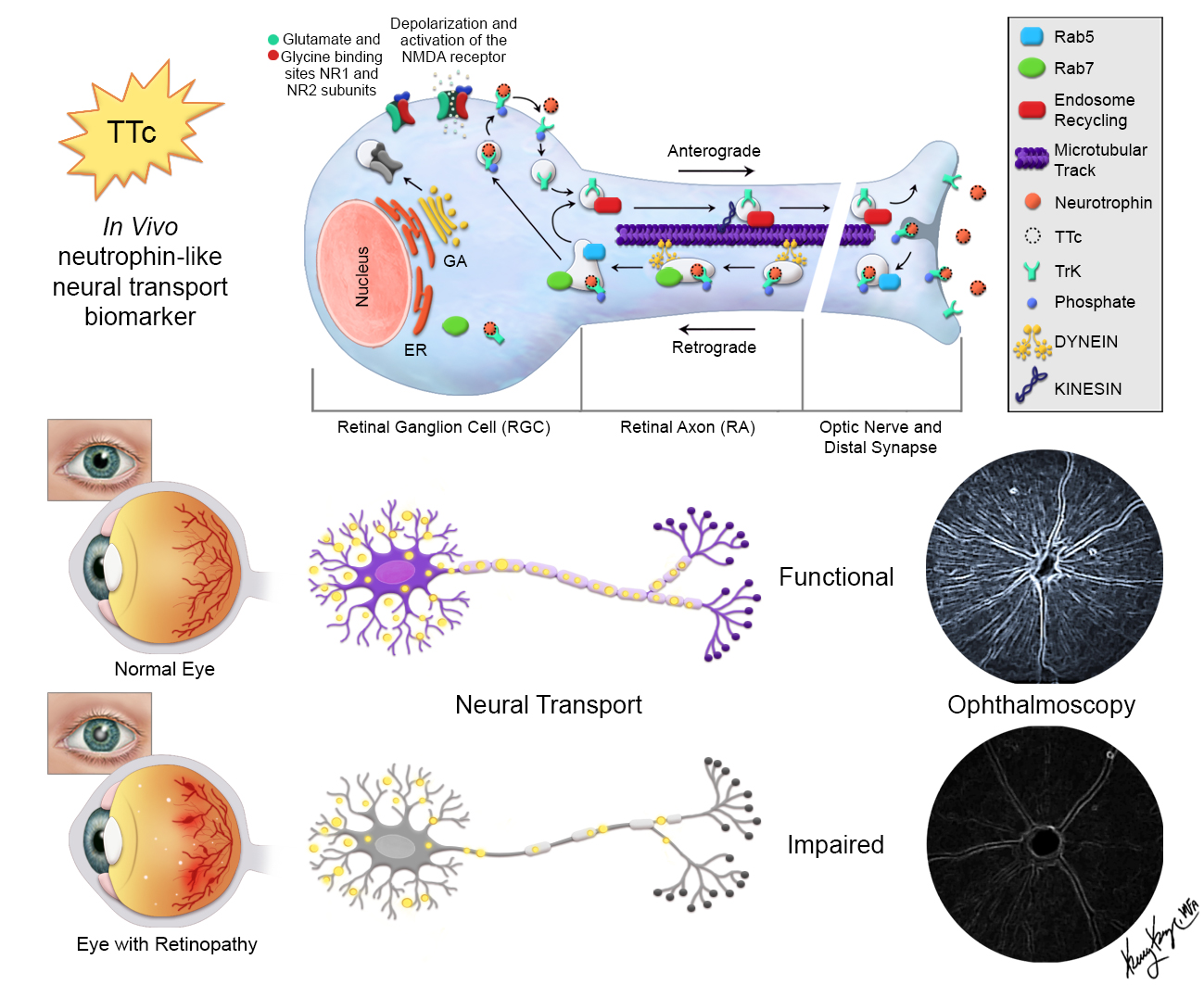
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Subjects
2.2. In Vivo Imaging
2.3. Ex Vivo Tissue Preparation
2.4. Immunofluorescence Histology
2.5. Ex Vivo Epifluorescence Imaging
2.6. ROIs and Transect Analysis
2.7. Ex Vivo Confocal Imaging
2.8. Statistical Analysis
3. Results
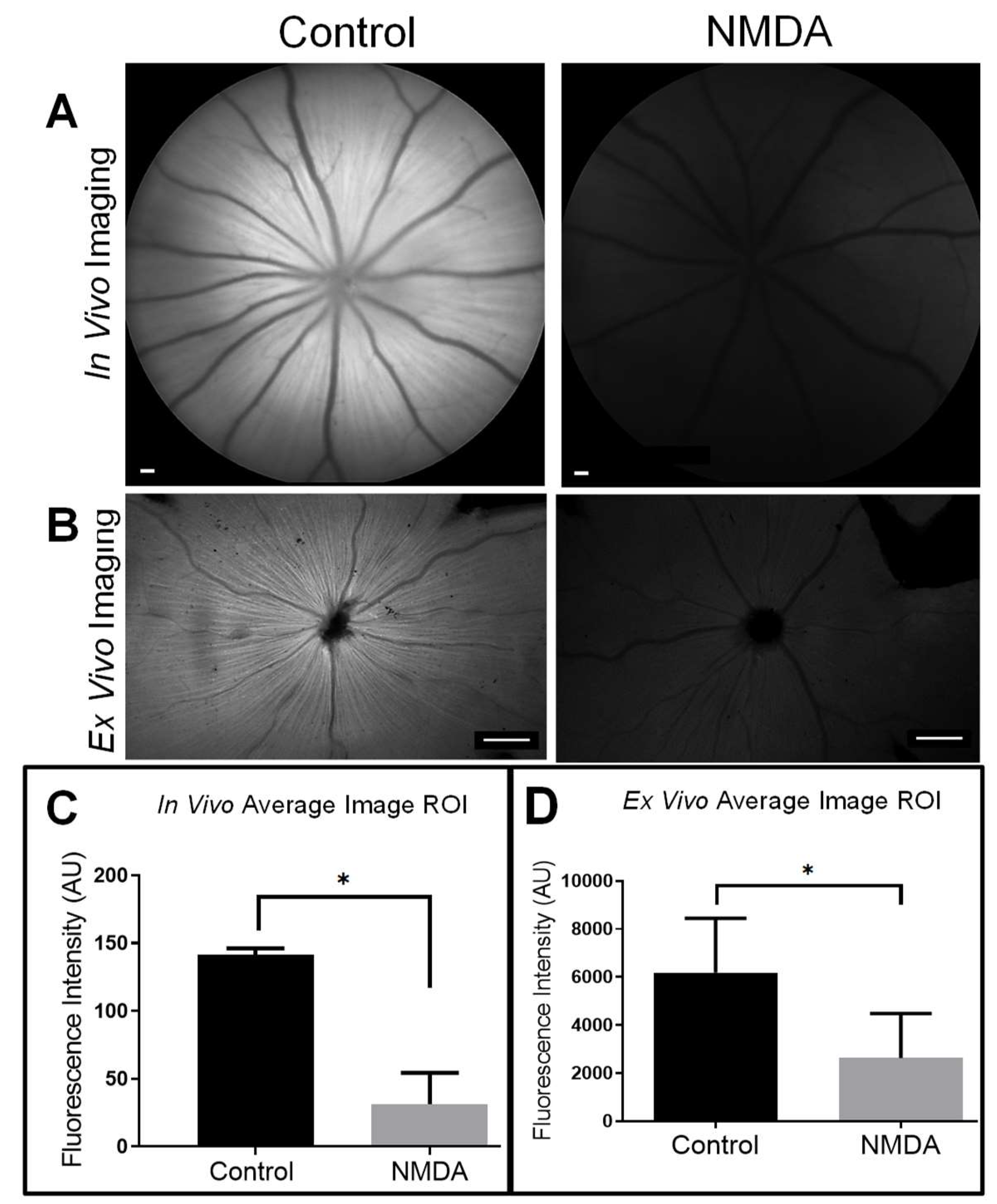
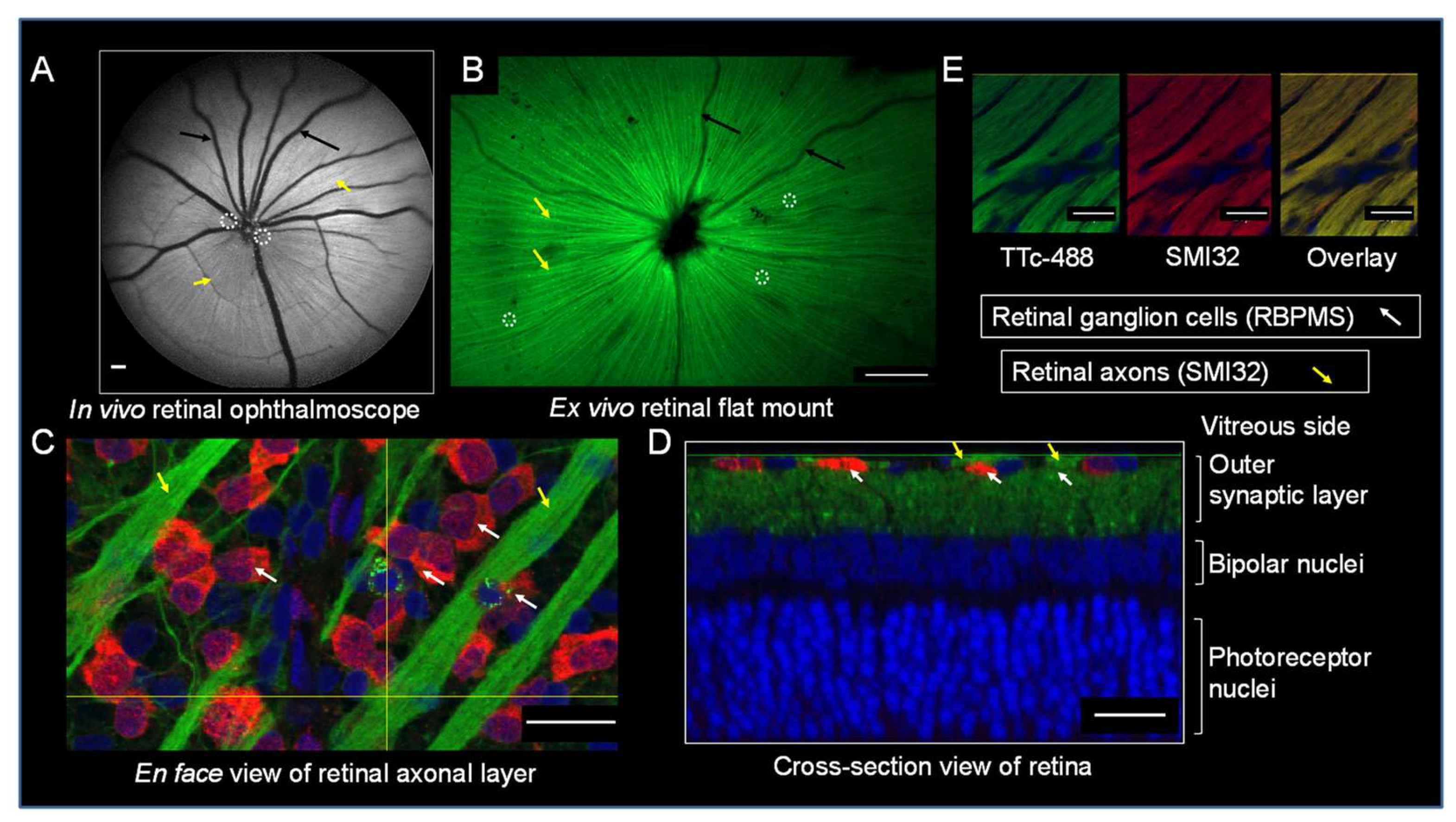
3.1. TTc is Rapidly Taken Up and Transported in Normal RGCs and RAs
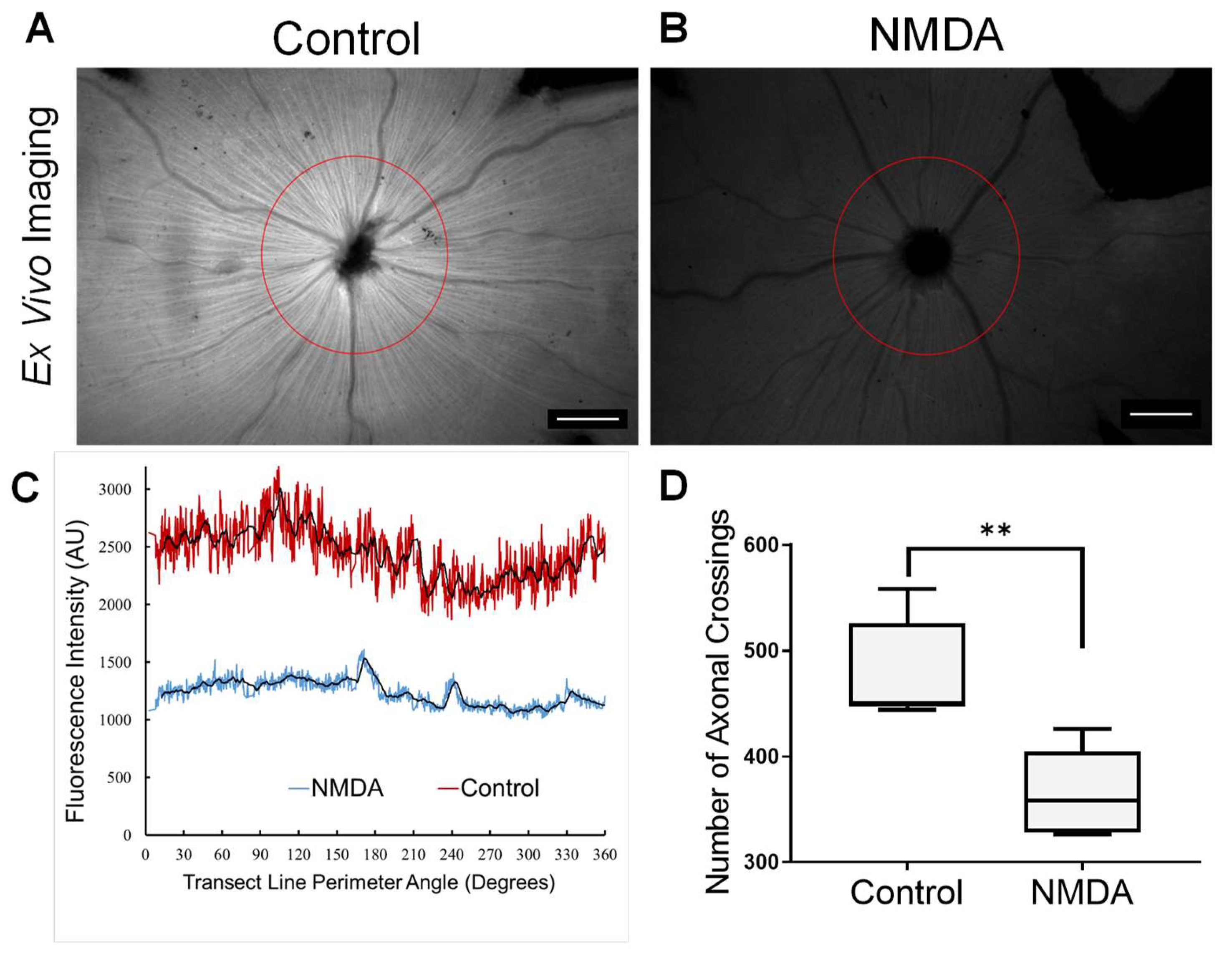
3.2. TTc Axonal Transport in a Retinopathic Model
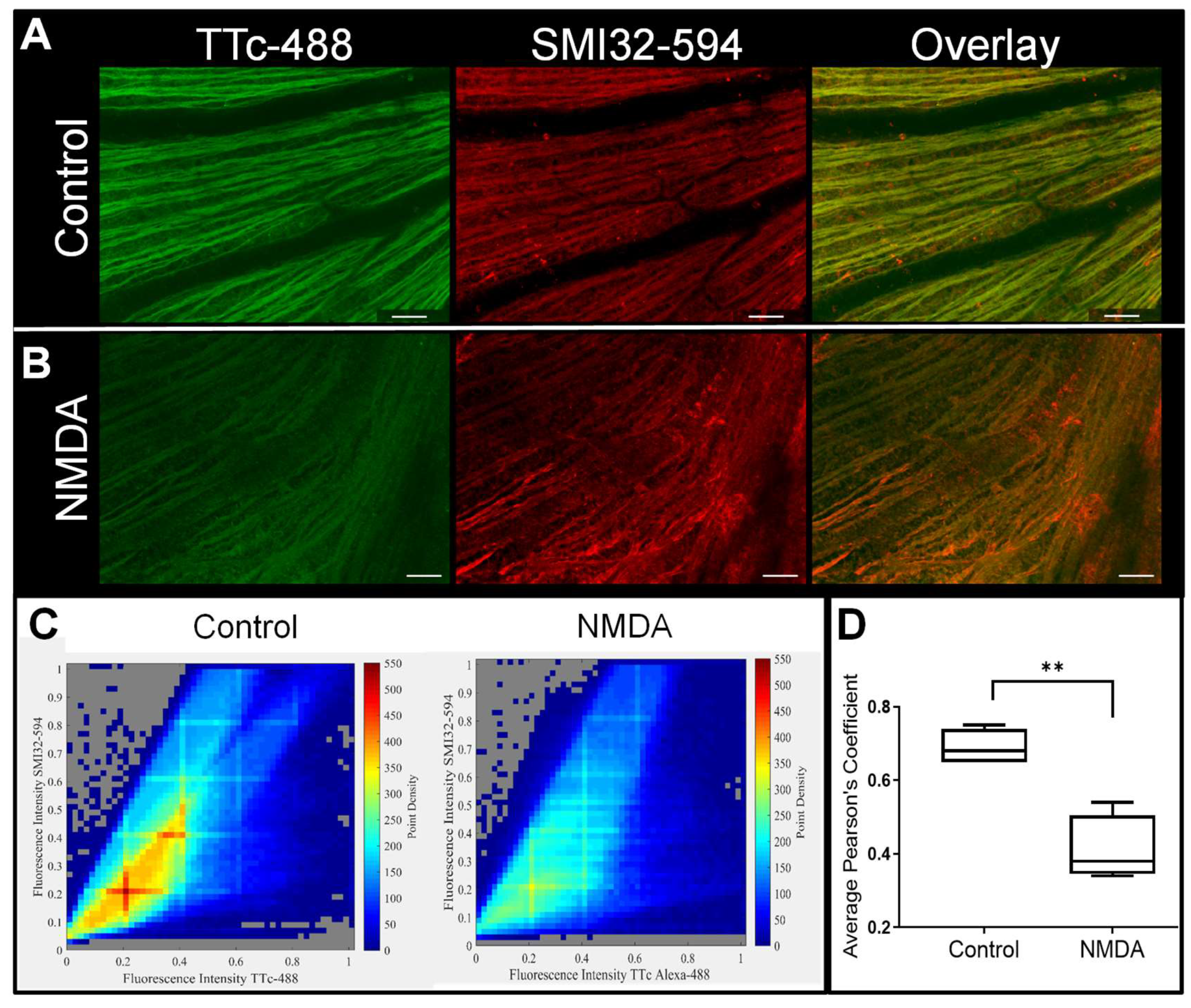
3.3. Retinal Axonal Damage and Axonal Transport in a Retinopathic Model
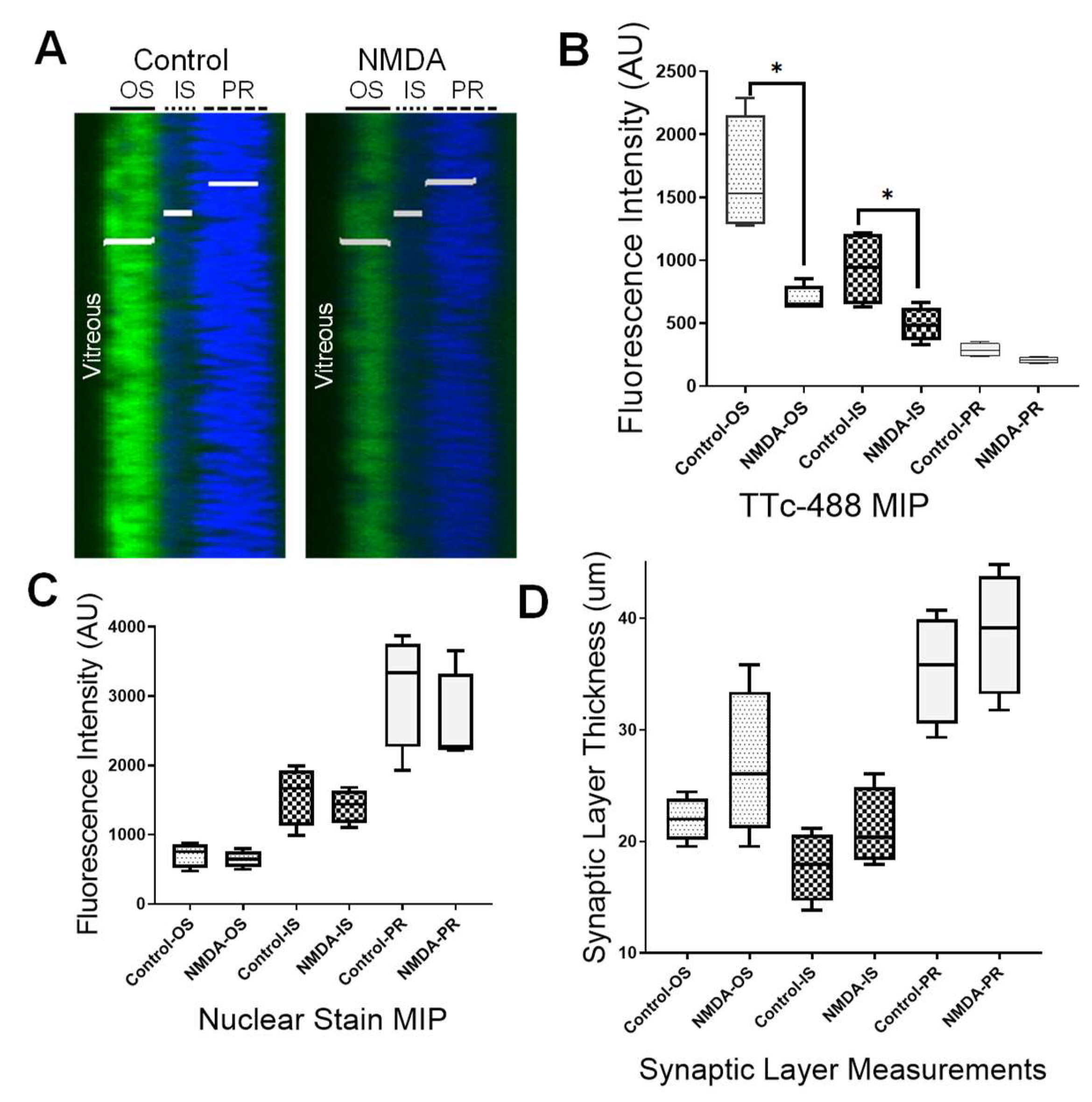
3.4. Retinal Synaptic Layers and Axonal Transport in a Retinopathic Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TTc | tetanus toxin C fragment |
| AMD | age related macular degeneration |
| RA | retinal axons |
| RGC | retinal ganglion cells |
| RNFL | retinal nerve fiber layer |
| CTB | cholera toxin B |
| CSLO | confocal scanning laser ophthalmoscope |
| RBPMS | RNA binding protein with multiple splicing |
| SMI32 | neurofilament marker |
| NMDA | N-methyl-D-aspartate |
| IF | immunofluorescence |
| SD | standard deviation |
| OS | outer synaptic layer |
| IS | inner synaptic layer |
| PS | photoreceptor layer |
| EX | excitation |
| EM | emission |
References
- National Eye Institute. Available online: https://nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases (accessed on 13 February 2020).
- Rein, D.B.; Zhang, P.; Wirth, K.E.; Lee, P.P.; Hoerger, T.J.; McCall, N.; Klein, R.; Tielsch, J.M.; Vijan, S.; Saaddine, J. The economic burden of major adult visual disorders in the United States. Arch. Ophthalmol. 2006, 124, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Rein, D.B.; Wirth, K.E.; Johnson, C.A.; Lee, P.P. Estimating quality-adjusted life year losses associated with visual field deficits using methodological approaches. Ophthalmic Epidemiol. 2007, 14, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Bourne, R.R.A.; Flaxman, S.R.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; Leasher, J.; Limburg, H.; et al. Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: a systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e888–e897. [Google Scholar] [CrossRef] [Green Version]
- Harwerth, R.S.; Quigley, H.A. Visual field defects and retinal ganglion cell losses in patients with glaucoma. Arch. Ophthalmol 2006, 124, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrigan-Baumrind, L.A.; Quigley, H.A.; Pease, M.E.; Kerrigan, D.F.; Mitchell, R.S. Number of ganglion cells in glaucoma eyes compared with threshold visual field tests in the same persons. Invest. Ophthalmol. Vis. Sci. 2000, 41, 741–748. [Google Scholar] [PubMed]
- Balendra, S.I.; Normando, E.M.; Bloom, P.A.; Cordeiro, M.F. Advances in retinal ganglion cell imaging. Eye 2015, 29, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Crabb, D.P.; Smith, N.D.; Glen, F.C.; Burton, R.; Garway-Heath, D.F. How does glaucoma look?: Patient perception of visual field loss. Ophthalmology 2013, 120, 1120–1126. [Google Scholar] [CrossRef]
- Thanos, S.; Indorf, L.; Naskar, R. In vivo FM: Using conventional fluorescence microscopy to monitor retinal neuronal death in vivo. Trends Neurosci. 2002, 25, 441–444. [Google Scholar] [CrossRef]
- Higashide, T.; Kawaguchi, I.; Ohkubo, S.; Takeda, H.; Sugiyama, K. In vivo imaging and counting of rat retinal ganglion cells using a scanning laser ophthalmoscope. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2943–2950. [Google Scholar] [CrossRef] [Green Version]
- Abbott, C.J.; Choe, T.E.; Lusardi, T.A.; Burgoyne, C.F.; Wang, L.; Fortune, B. Imaging axonal transport in the rat visual pathway. Biomed. Opt.Express. 2013, 4, 364–386. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Johnson, J.R.; Wilson, B.S.; Gammon, S.T.; Piwnica-Worms, D.; Barnett, E.M. Single-cell resolution imaging of retinal ganglion cell apoptosis in vivo using a cell-penetrating caspase-activatable peptide probe. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, E.M.; Zhang, X.; Maxwell, D.; Chang, Q.; Piwnica-Worms, D. Single-cell imaging of retinal ganglion cell apoptosis with a cell-penetrating, activatable peptide probe in an in vivo glaucoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 9391–9896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, D.; Chang, Q.; Zhang, X.; Barnett, E.M.; Piwnica-Worms, D. An improved cell-penetrating, caspase-activatable, near-infrared fluorescent peptide for apoptosis imaging. Bioconjug. Chem. 2009, 20, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, M.F.; Migdal, C.; Bloom, P.; Fitzke, F.W.; Moss, S.E. Imaging apoptosis in the eye. Eye 2011, 25, 545–553. [Google Scholar] [CrossRef]
- Quigley, H.A.; Addicks, E.M. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest. Ophthalmol. Vis. Sci. 1980, 19, 137–152. [Google Scholar]
- Minckler, D.S.; Bunt, A.H.; Johanson, G.W. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest. Ophthalmol. Vis. Sci. 1977, 16, 426–441. [Google Scholar]
- Salinas-Navarro, M.; Alarcon-Martinez, L.; Valiente-Soriano, F.J.; Jimenez-Lopez, M.; Mayor-Torroglosa, S.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M. Ocular hypertension impairs optic nerve axonal transport leading to progressive retinal ganglion cell degeneration. Exp Eye Res 2010, 90, 168–183. [Google Scholar] [CrossRef]
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef]
- Morgan, J.E. Circulation and axonal transport in the optic nerve. Eye 2004, 18, 1089–1095. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Roy, S.; Zhang, B.; Lee, V.M.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta. Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Perlson, E.; Maday, S.; Fu, M.M.; Moughamian, A.J.; Holzbaur, E.L. Retrograde axonal transport: Pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieg, N.; Menendez, G.; Schiavo, G.; Terenzio, M. Signaling endosomes in axonal transport: travel updates on the molecular highway. Semin. Cell Dev. Biol. 2014, 27, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Terenzio, M.; Schiavo, G.; Fainzilber, M. Compartmentalized signaling in neurons: From cell biology to neuroscience. Neuron 2017, 96, 667–679. [Google Scholar] [CrossRef]
- Surana, S.; Tosolini, A.P.; Meyer, I.F.G.; Fellows, A.D.; Novoselov, S.S.; Schiavo, G. The travel diaries of tetanus and botulinum neurotoxins. Toxicon 2018, 147, 58–67. [Google Scholar] [CrossRef]
- Lam, T.T.; Abler, A.S.; Kwong, J.M.; Tso, M.O. N-methyl-D-aspartate (NMDA)-induced apoptosis in rat retina. Invest. Ophthalmol. Vis. Sci. 1999, 40, 2391–2397. [Google Scholar]
- McKinnon, S.J.; Schlamp, C.L.; Nickells, R.W. Mouse models of retinal ganglion cell death and glaucoma. Exp. Eye Res. 2009, 88, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Lim, J.K.; Li, Q.X.; He, Z.; Vingrys, A.J.; Wong, V.H.; Currier, N.; Mullen, J.; Bui, B.V.; Nguyen, C.T. The eye as a biomarker for Alzheimer’s disease. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Normando, E.M.; Davis, B.M.; De Groef, L.; Nizari, S.; Turner, L.A.; Ravindran, N.; Pahlitzsch, M.; Brenton, J.; Malaguarnera, G.; Guo, L.; et al. The retina as an early biomarker of neurodegeneration in a rotenone-induced model of Parkinson’s disease: Evidence for a neuroprotective effect of rosiglitazone in the eye and brain. Acta Neuropathol. Commun. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Raff, M.C.; Whitmore, A.V.; Finn, J.T. Axonal self-destruction and neurodegeneration. Science 2002, 296, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.P.; Perry, V.H. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002, 25, 532–537. [Google Scholar] [CrossRef]
- Whitmore, A.V.; Libby, R.T.; John, S.W. Glaucoma: Thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog. Retin. Eye Res. 2005, 24, 639–662. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, B.P.; Inman, D.M.; Lambert, W.; Oglesby, E.; Calkins, D.J.; Steele, M.R.; Vetter, M.L.; Marsh-Armstrong, N.; Horner, P.J. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J. Neurosci. 2008, 28, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzini, B.; Grob, P.; Akert, K. Papain-derived fragment IIc of tetanus toxin: Its binding to isolated synaptic membranes and retrograde axonal transport. Brain Res 1981, 210, 291–299. [Google Scholar] [CrossRef]
- Fishman, P.S.; Savitt, J.M. Transsynaptic transfer of retrogradely transported tetanus protein-peroxidase conjugates. Exp. Neurol. 1989, 106, 197–203. [Google Scholar] [CrossRef]
- Fishman, P.S.; Savitt, J.M.; Farrand, D.A. Enhanced CNS uptake of systemically administered proteins through conjugation with tetanus C-fragment. J. Neurol. Sci. 1990, 98, 311–325. [Google Scholar] [CrossRef]
- Herreros, J.; Lalli, G.; Schiavo, G. C-terminal half of tetanus toxin fragment C is sufficient for neuronal binding and interaction with a putative protein receptor. Biochem. J. 2000, 347, 199–204. [Google Scholar] [CrossRef]
- Lalli, G.; Gschmeissner, S.; Schiavo, G. Myosin Va and microtubule-based motors are required for fast axonal retrograde transport of tetanus toxin in motor neurons. J. Cell. Sci. 2003, 116, 4639–4650. [Google Scholar] [CrossRef] [Green Version]
- Herreros, J.; Ng, T.; Schiavo, G. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol. Biol. Cell 2001, 12, 2947–2960. [Google Scholar] [CrossRef] [Green Version]
- Deinhardt, K.; Berninghausen, O.; Willison, H.J.; Hopkins, C.R.; Schiavo, G. Tetanus toxin is internalized by a sequential clathrin-dependent mechanism initiated within lipid microdomains and independent of epsin1. J. Cell. Biol. 2006, 174, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell. Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Beaude, P.; Delacour, A.; Bizzini, B.; Domuado, D.; Remy, M.H. Retrograde axonal transport of an exogenous enzyme covalently linked to B-IIb fragment of tetanus toxin. Biochem. J. 1990, 271, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Carlton, E.; Teng, Q.; Federici, T.; Yang, J.; Riley, J.; Boulis, N.M. Fusion of the tetanus toxin C fragment binding domain and Bcl-xL for protection of peripheral nerve neurons. Neurosurgery 2008, 63, 1175–1182. [Google Scholar] [CrossRef]
- Ciriza, J.; Moreno-Igoa, M.; Calvo, A.C.; Yague, G.; Palacio, J.; Miana-Mena, F.J.; Munoz, M.J.; Zaragoza, P.; Brulet, P.; Osta, R. A genetic fusion GDNF-C fragment of tetanus toxin prolongs survival in a symptomatic mouse ALS model. Restor. Neurol. Neurosci. 2008, 26, 459–465. [Google Scholar]
- Le Roux, L.G.; Schellingerhout, D.; Hobbs, B.P.; Bredow, S. Impairment of retrograde neuronal transport in oxaliplatin-induced neuropathy demonstrated by molecular imaging. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Le Roux, L.G.; Bredow, S.; Grosshans, D.; Schellingerhout, D. Molecular imaging detects impairment in the retrograde axonal transport mechanism after radiation-induced spinal cord injury. Mol. Imaging Biol. 2014, 16, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Bizzini, B.; Stoeckel, K.; Schwab, M. An antigenic polypeptide fragment isolated from tetanus toxin: Chemical characterization, binding to gangliosides and retrograde axonal transport in various neuron systems. J. Neurochem. 1977, 28, 529–542. [Google Scholar] [CrossRef]
- Evinger, C.; Erichsen, J.T. Transsynaptic retrograde transport of fragment C of tetanus toxin demonstrated by immunohistochemical localization. Brain Res. 1986, 380, 383–388. [Google Scholar] [CrossRef]
- Mellanby, J.; Green, J. How does tetanus toxin act? Neuroscience 1981, 6, 281–300. [Google Scholar] [CrossRef]
- Manning, K.A.; Erichsen, J.T.; Evinger, C. Retrograde transneuronal transport properties of fragment C of tetanus toxin. Neuroscience 1990, 34, 251–263. [Google Scholar] [CrossRef]
- Nadal-Nicolas, F.M.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo-Barriuso, M. Two methods to trace retinal ganglion cells with fluorogold: From the intact optic nerve or by stereotactic injection into the optic tract. Exp. Eye Res. 2015, 131, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.X.; Yang, J.; Yuan, T.F.; So, K.F. Uptake of retrograde tracers by intact optic nerve axons: A new way to label retinal ganglion cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Villena, A.; Moreno, M.; Vidal, L.; Parrado, C.; Garcia-Campos, J.; de Vargas, I.P. Effects of a non-selective beta-blocker on adult rat anterograde axonal transport and retinal ganglion layer after increased intraocular pressure. Histol. Histopathol. 2005, 20, 1077–1084. [Google Scholar]
- Wang, X.; Baldridge, W.H.; Chauhan, B.C. Acute endothelin-1 application induces reversible fast axonal transport blockade in adult rat optic nerve. Invest. Ophthalmol. Vis. Sci. 2008, 49, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Hoffmann, K.P. Retinal projections to the pretectum, accessory optic system and superior colliculus in pigmented and albino ferrets. Eur. J. Neurosci. 1993, 5, 486–500. [Google Scholar] [CrossRef]
- Miceli, D.; Reperant, J.; Marchand, L.; Rio, J.P. Retrograde transneuronal transport of the fluorescent dye rhodamine beta-isothiocyanate from the primary and centrifugal visual systems in the pigeon. Brain Res. 1993, 601, 289–298. [Google Scholar] [CrossRef]
- Niwa, M.; Aoki, H.; Hirata, A.; Tomita, H.; Green, P.G.; Har, A. Retinal cell degeneration in animal models. Int. J. Mol. Sci. 2016, 17, 1–16. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Roux, L.G.; Qiu, X.; Jacobsen, M.C.; Pagel, M.D.; Gammon, S.T.; Piwnica-Worms, D.R.; Schellingerhout, D. Axonal Transport as an In Vivo Biomarker for Retinal Neuropathy. Cells 2020, 9, 1298. https://doi.org/10.3390/cells9051298
Le Roux LG, Qiu X, Jacobsen MC, Pagel MD, Gammon ST, Piwnica-Worms DR, Schellingerhout D. Axonal Transport as an In Vivo Biomarker for Retinal Neuropathy. Cells. 2020; 9(5):1298. https://doi.org/10.3390/cells9051298
Chicago/Turabian StyleLe Roux, Lucia G., Xudong Qiu, Megan C. Jacobsen, Mark D. Pagel, Seth T. Gammon, David R. Piwnica-Worms, and Dawid Schellingerhout. 2020. "Axonal Transport as an In Vivo Biomarker for Retinal Neuropathy" Cells 9, no. 5: 1298. https://doi.org/10.3390/cells9051298
APA StyleLe Roux, L. G., Qiu, X., Jacobsen, M. C., Pagel, M. D., Gammon, S. T., Piwnica-Worms, D. R., & Schellingerhout, D. (2020). Axonal Transport as an In Vivo Biomarker for Retinal Neuropathy. Cells, 9(5), 1298. https://doi.org/10.3390/cells9051298