Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis

 ,
,  , ,
, ,
Abstract
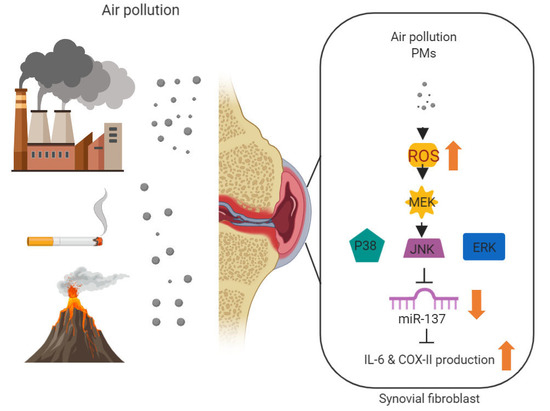
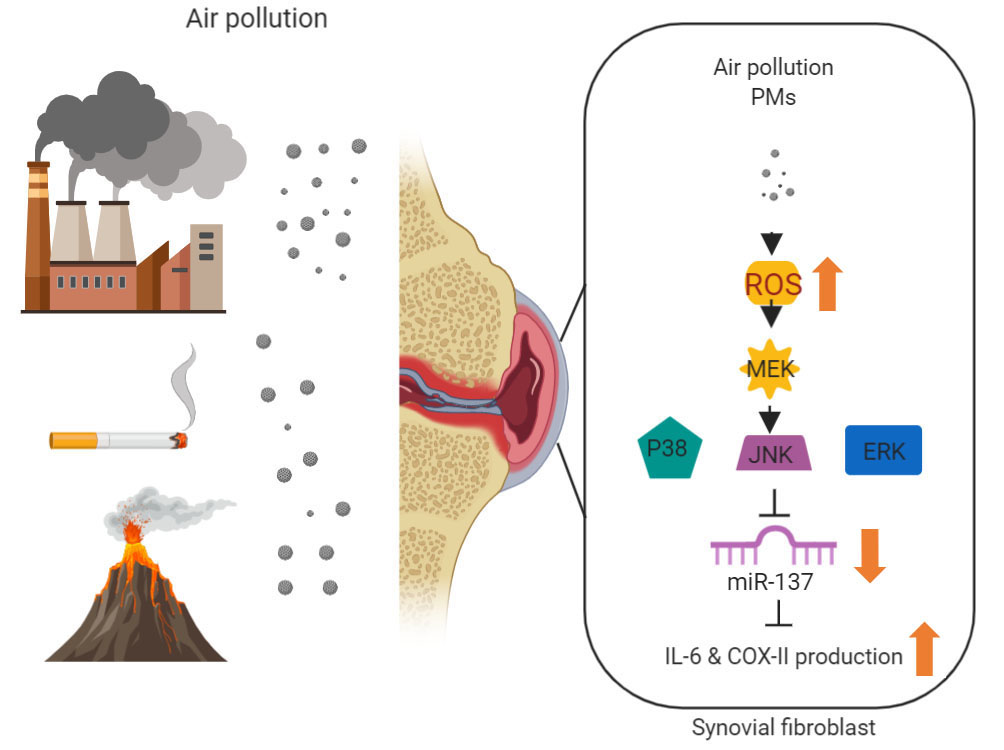
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
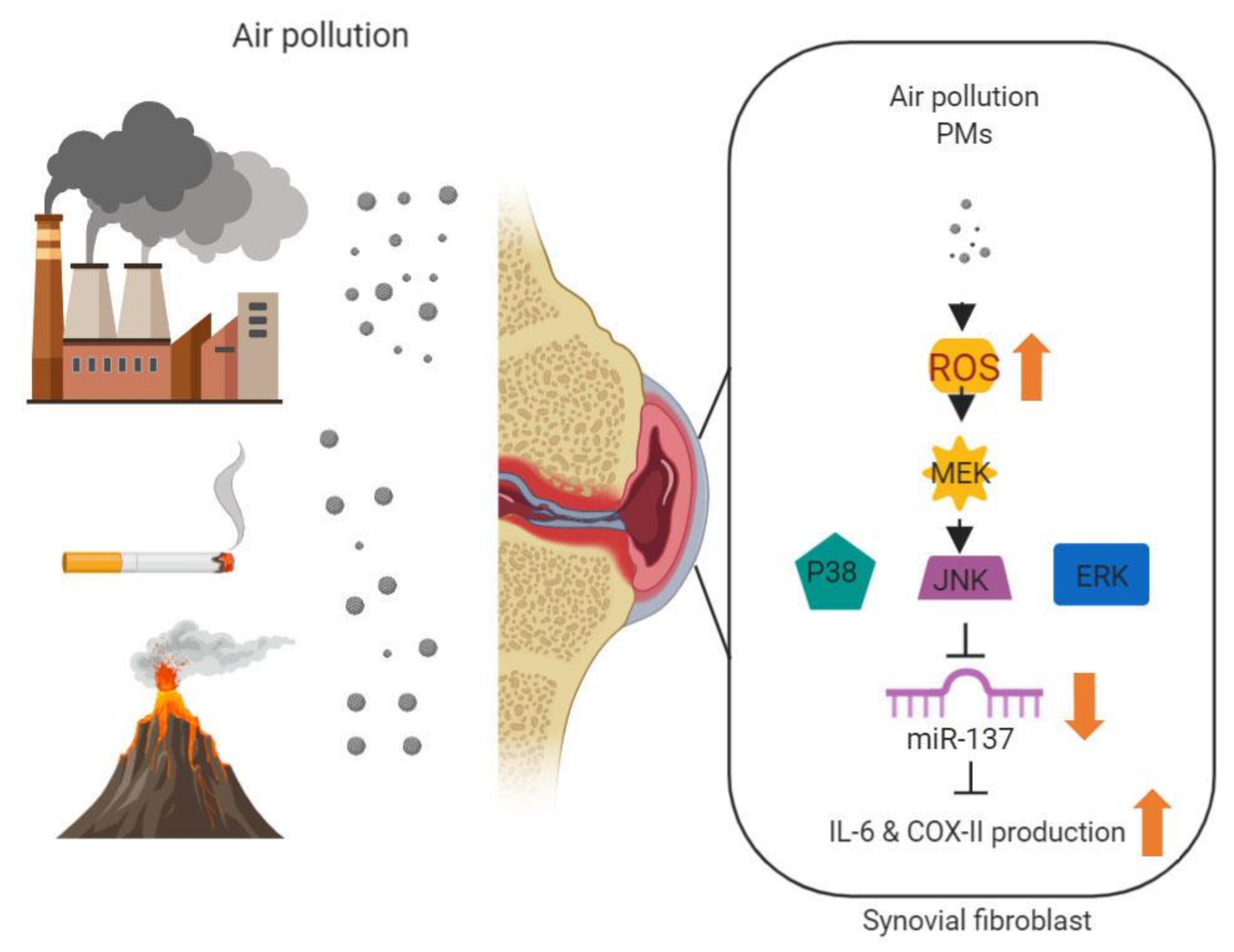{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Human Synovial Fluids and Tissues
- (1)
- Patients who were able and willing to provide written informed consent;
- (2)
- Patients who had sufficient knowledge to understand Chinese or Taiwanese language, so they could comply with the requirements of the study;
- (3)
- Patients who were at least 18 years old, but less than 90 years old;
- (4)
- Patients who were diagnosed as having RA by their rheumatologist and met the 2010 ACR/EULAR classification criteria for RA (Aletaha D, et al., 2010);
- (5)
- Patients who had active RA defined as a clinical disease activity index (CDAI) > 10 and had a swollen knee joint;
- (1)
- Patients who had any other inflammatory rheumatic disease than RA, including secondary Sjögren’s syndrome;
- (2)
- Patients who refused to sign or who could not understand either Chinese or Taiwanese;
- (3)
- Patients who could not tolerate the procedure of knee treatment.
- (4)
- Patients who were pregnant.
2.3. Preparation of Particle Matter Samples
2.4. Transient Transfection
2.5. Reporter Gene Assay
2.6. Plasmid Construction and Luciferase Assays
2.7. Collagen-Induced Arthritis (CIA) Rat Model
2.8. Immunohistochemistry
2.9. Statistical Analysis
3. Results
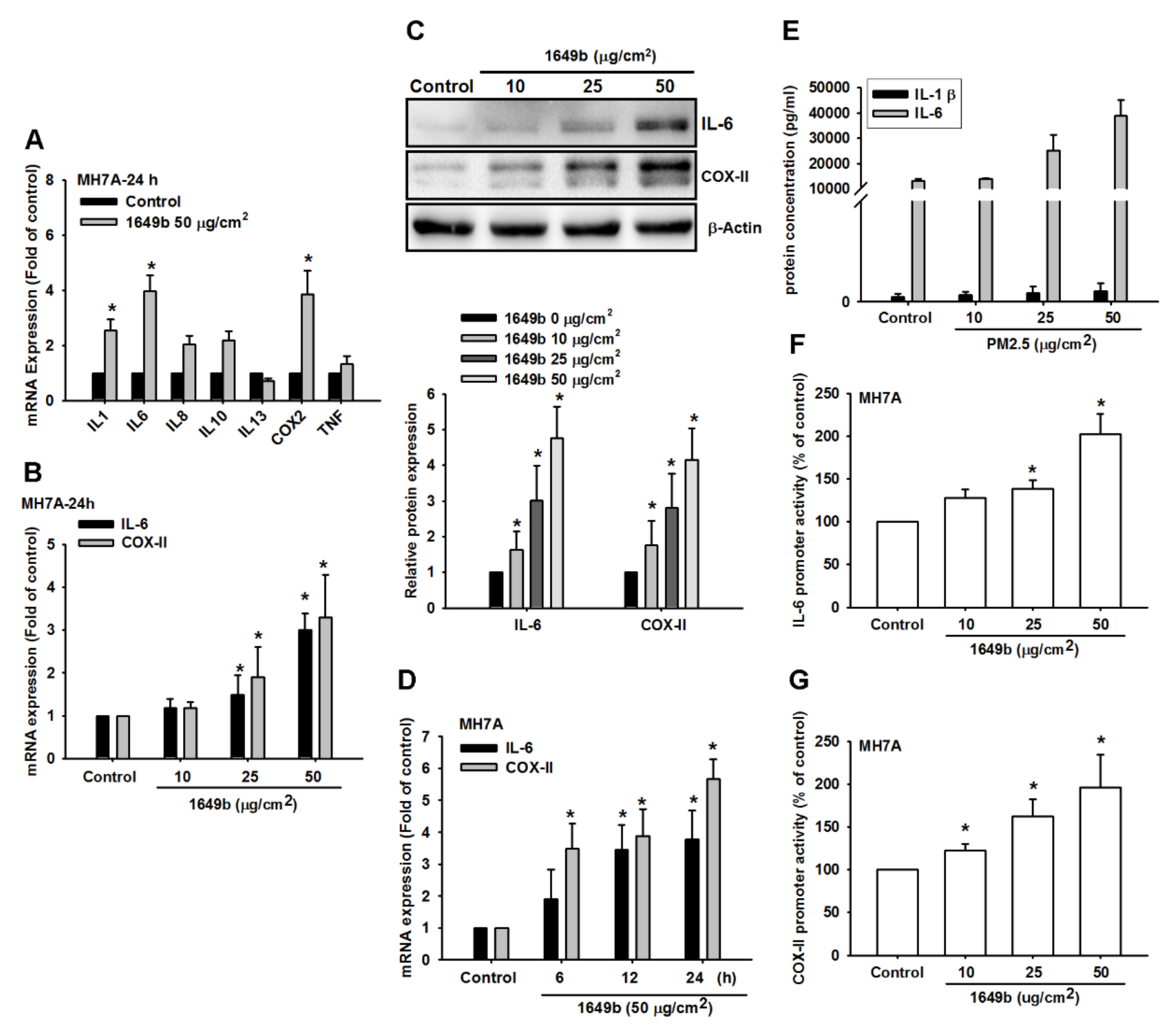
3.1. PMs Promote IL-6 and COX-II Expression in Human Rheumatoid Arthritis Fibroblast-Like Synoviocytes (RA-FLS)
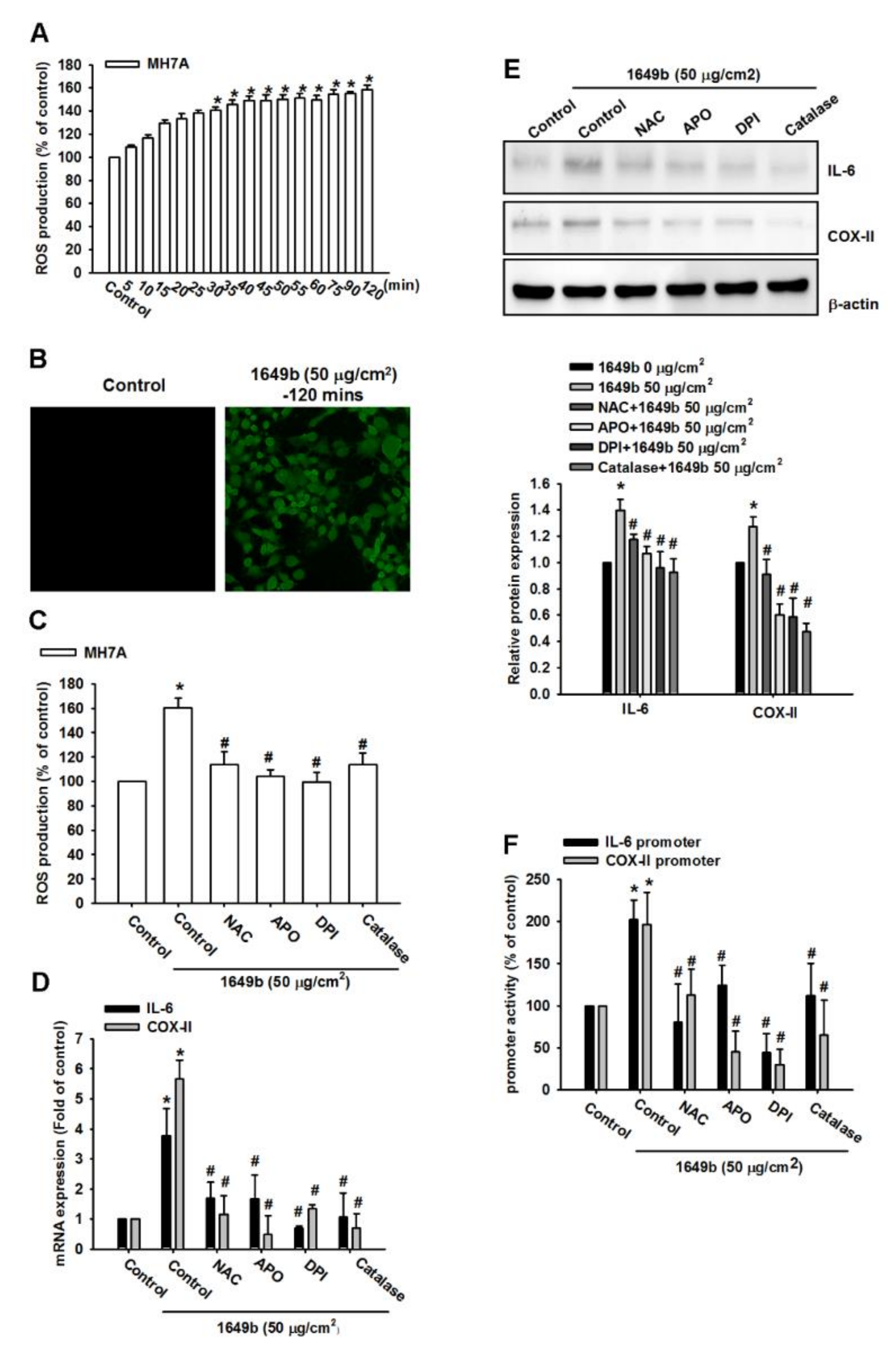
3.2. PMs Induces IL-6 and COX-II Expression via ROS
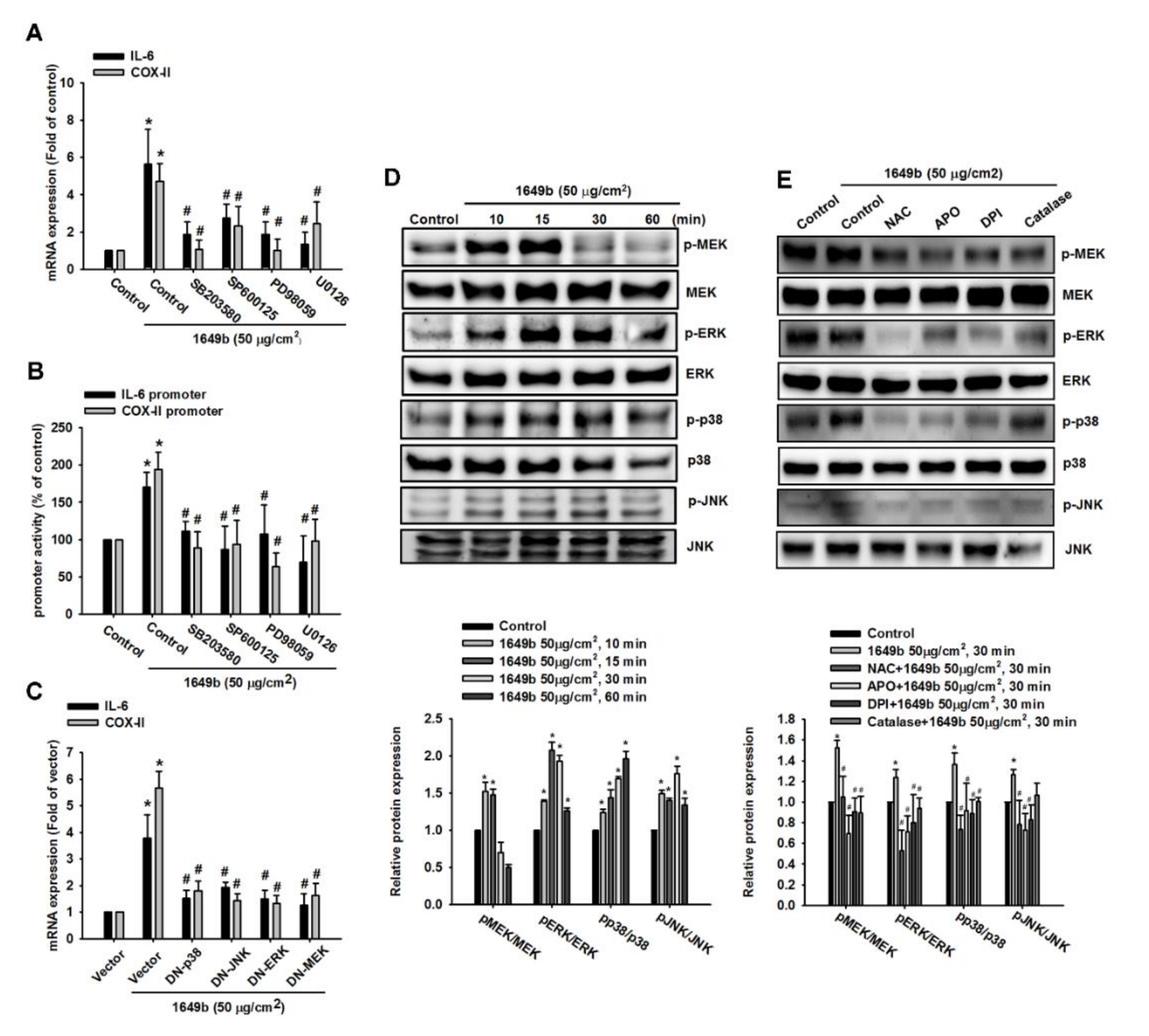
3.3. PMs Induce IL-6 and COX-II Expression via MAPK in Human RA-FLS
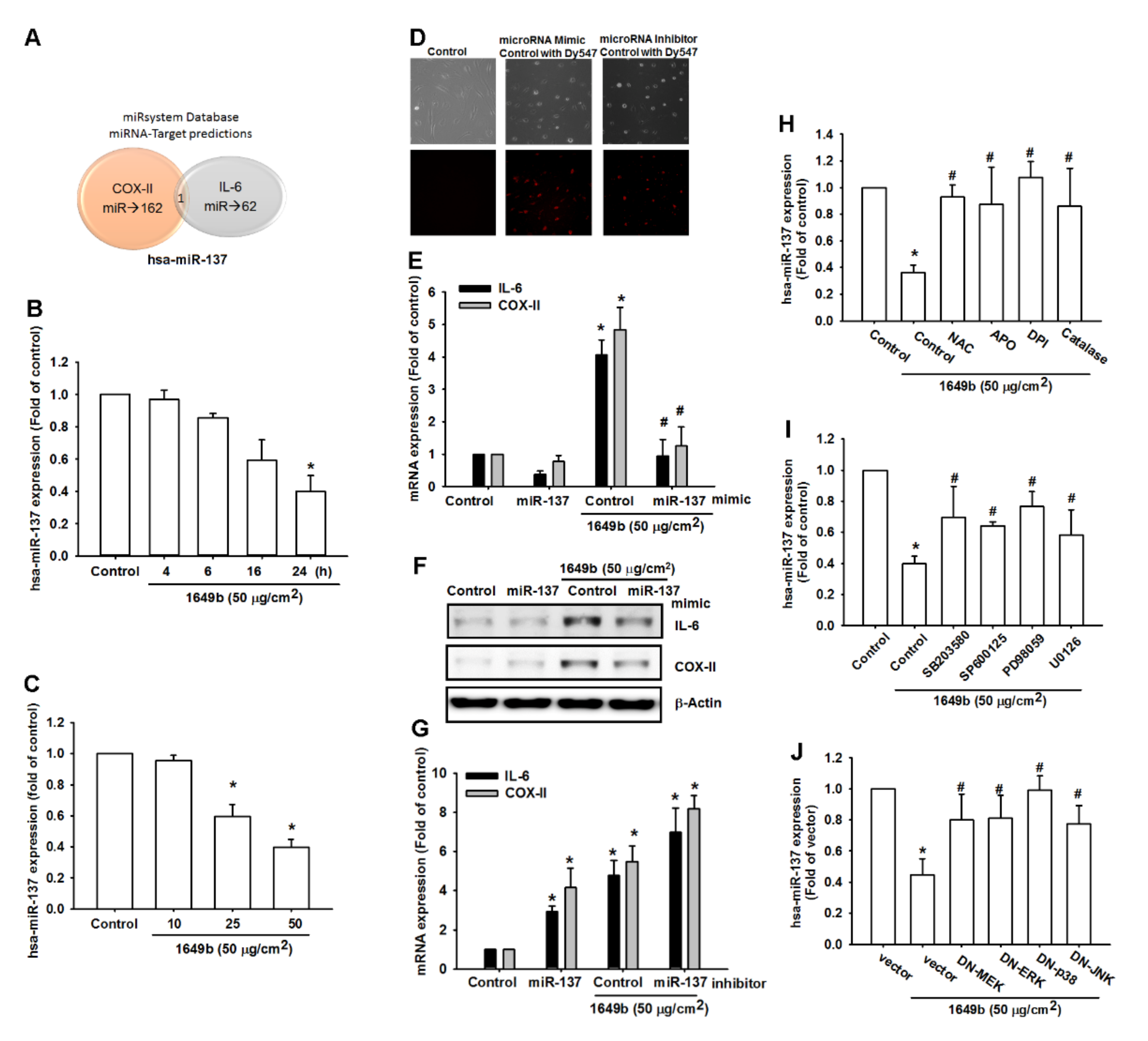
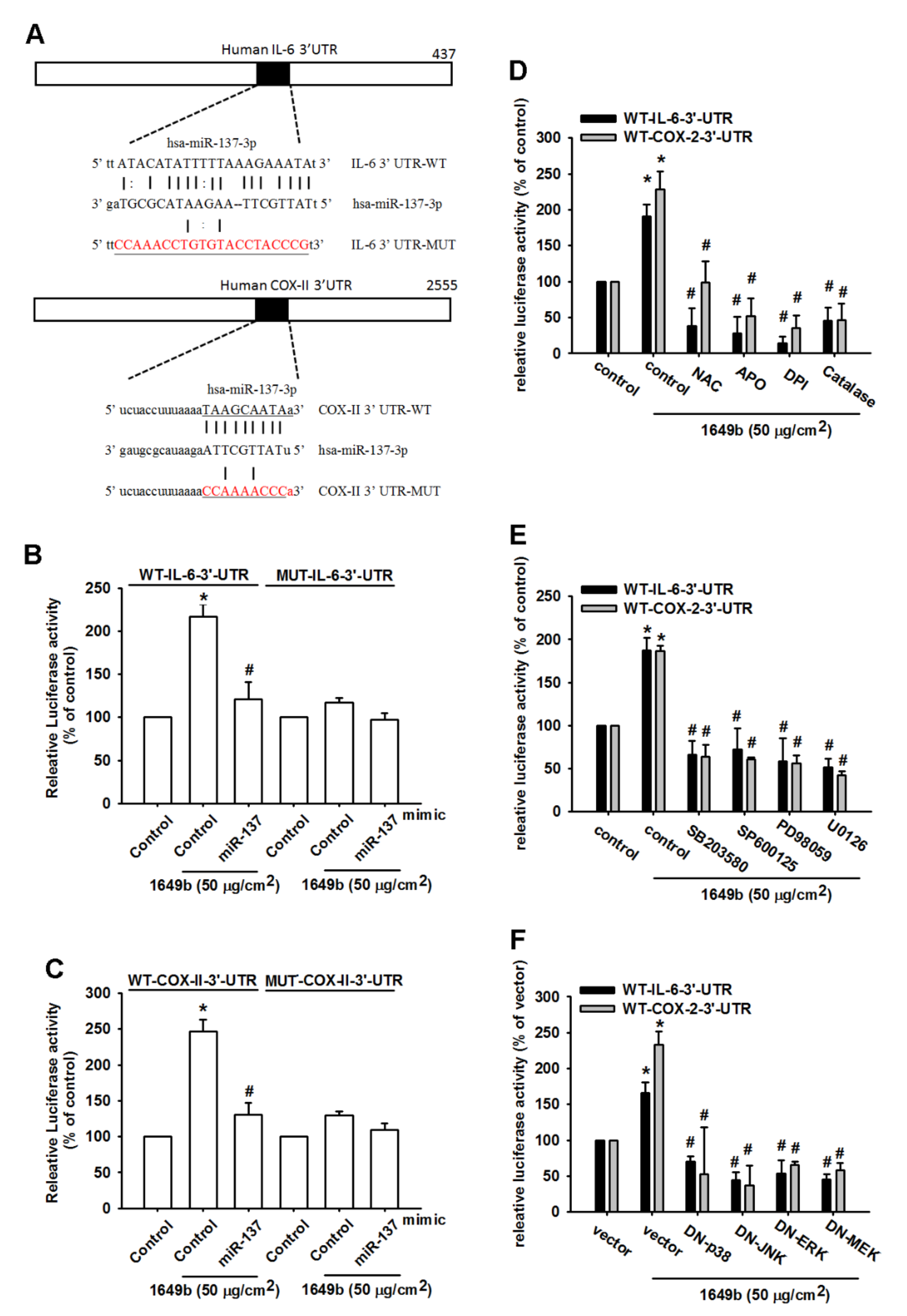
3.4. PMs Enhances IL-6 and COX-II Expression by Inhibiting Hsa-miR-137 Synthesis
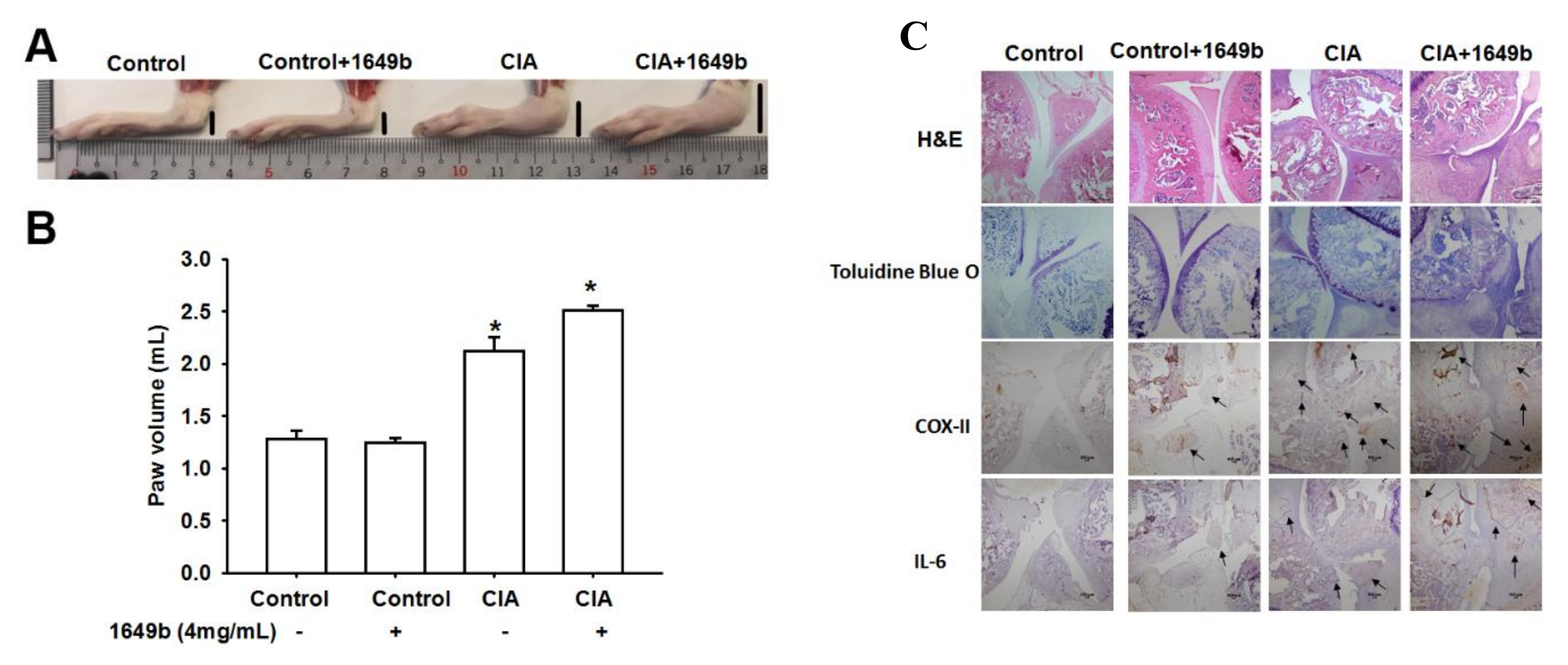
3.5. PMs Enhanced CIA-Induced Arthritis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Ethics Approval and Consent to Participate
Conflicts of Interest
References
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, N.; Pascal, V.; Fasth, A.E.; Sundstrom, Y.; Galsgaard, E.D.; Ahern, D.; Andersen, M.; Baslund, B.; Bartels, E.M.; Bliddal, H.; et al. Balance between activating NKG2D, DNAM-1, NKp44 and NKp46 and inhibitory CD94/NKG2A receptors determine natural killer degranulation towards rheumatoid arthritis synovial fibroblasts. Immunology 2014, 142, 581–593. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Buckley, C.D.; Isaacs, J.D. Cytokines in rheumatoid arthritis-shaping the immunological landscape. Nat. Rev. Rheumatol. 2016, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Furst, D.E.; Emery, P. Rheumatoid arthritis pathophysiology: Update on emerging cytokine and cytokine-associated cell targets. Rheumatology 2014, 53, 1560–1569. [Google Scholar] [CrossRef] [Green Version]
- Moran-Moguel, M.C.; Petarra-Del Rio, S.; Mayorquin-Galvan, E.E.; Zavala-Cerna, M.G. Rheumatoid Arthritis and miRNAs: A Critical Review through a Functional View. J. Immunol. Res 2018, 2018, 2474529. [Google Scholar] [CrossRef]
- Filkova, M.; Aradi, B.; Senolt, L.; Ospelt, C.; Vettori, S.; Mann, H.; Filer, A.; Raza, K.; Buckley, C.D.; Snow, M.; et al. Association of circulating miR-223 and miR-16 with disease activity in patients with early rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 1898–1904. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, K.; Kochi, Y.; Ikari, K.; Yamamoto, K.; Suzuki, A.; Shimane, K.; Nakamura, Y.; Yano, K.; Iikuni, N.; Tsukahara, S.; et al. Association study of TRAF1-C5 polymorphisms with susceptibility to rheumatoid arthritis and systemic lupus erythematosus in Japanese. Ann. Rheum. Dis. 2010, 69, 368–373. [Google Scholar] [CrossRef]
- Chatzikyriakidou, A.; Voulgari, P.V.; Georgiou, I.; Drosos, A.A. miRNAs and related polymorphisms in rheumatoid arthritis susceptibility. Autoimmun. Rev. 2012, 11, 636–641. [Google Scholar] [CrossRef]
- Jung, C.R.; Hsieh, H.Y.; Hwang, B.F. Air Pollution as a Potential Determinant of Rheumatoid Arthritis: A Population-based Cohort Study in Taiwan. Epidemiology 2017, 28 (Suppl. 1), S54–S59. [Google Scholar] [CrossRef]
- Too, C.L.; Muhamad, N.A.; Ilar, A.; Padyukov, L.; Alfredsson, L.; Klareskog, L.; Murad, S.; Bengtsson, C.; My, E.S.G. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: Results from a Malaysian population-based case-control study. Ann. Rheum. Dis. 2016, 75, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.E.; Kallberg, H.; Laden, F.; Bellander, T.; Costenbader, K.H.; Holmqvist, M.; Klareskog, L.; Alfredsson, L.; Karlson, E.W. Ambient air pollution exposures and risk of rheumatoid arthritis: Results from the Swedish EIRA case-control study. Ann. Rheum. Dis. 2013, 72, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Farhat, S.C.; Silva, C.A.; Orione, M.A.; Campos, L.M.; Sallum, A.M.; Braga, A.L. Air pollution in autoimmune rheumatic diseases: A review. Autoimmun. Rev. 2011, 11, 14–21. [Google Scholar] [CrossRef]
- Ospelt, C.; Bang, H.; Feist, E.; Camici, G.; Keller, S.; Detert, J.; Kramer, A.; Gay, S.; Ghannam, K.; Burmester, G.R. Carbamylation of vimentin is inducible by smoking and represents an independent autoantigen in rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1176–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, G.; Xia, J.; Zhang, Y.; Guo, L.; Chen, R.; Sang, N. Ambient fine particulate matter exposure induces reversible cardiac dysfunction and fibrosis in juvenile and older female mice. Part. Fibre Toxicol. 2018, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.V.; Yu, J.; Guo, Y.; Byun, J.; Chen, Y.E.; Wang, L.; Liu, M.; Bard, R.L.; Morishita, M.; Huang, W.; et al. Effect of Ambient Fine Particulate Matter Air Pollution and Colder Outdoor Temperatures on High-Density Lipoprotein Function. Am. J. Cardiol. 2018, 122, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.P.; Li, Z.; Choi, E.K.; Lee, S.; Kim, Y.K.; Seo, E.Y.; Chung, J.H.; Cho, S. Urban particulate matter in air pollution penetrates into the barrier-disrupted skin and produces ROS-dependent cutaneous inflammatory response in vivo. J. Dermatol. Sci. 2018, 91, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ji, C.; Huang, Y.; Aniagu, S.; Jiang, Y.; Chen, T. AHR-mediated ROS production contributes to the cardiac developmental toxicity of PM2.5 in zebrafish embryos. Sci. Total Environ. 2020, 719, 135097. [Google Scholar] [CrossRef] [PubMed]
- Ovrevik, J. Oxidative Potential Versus Biological Effects: A Review on the Relevance of Cell-Free/Abiotic Assays as Predictors of Toxicity from Airborne Particulate Matter. Int. J. Mol. Sci 2019, 20, 4772. [Google Scholar] [CrossRef] [Green Version]
- Niu, Q.; Huang, Z.C.; Wu, X.J.; Jin, Y.X.; An, Y.F.; Li, Y.M.; Xu, H.; Yang, B.; Wang, L.L. Enhanced IL-6/phosphorylated STAT3 signaling is related to the imbalance of circulating T follicular helper/T follicular regulatory cells in patients with rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 200. [Google Scholar] [CrossRef] [Green Version]
- Niu, Q.; Cai, B.; Huang, Z.C.; Shi, Y.Y.; Wang, L.L. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol. Int 2012, 32, 2731–2736. [Google Scholar] [CrossRef]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Hsu, L.F.; Lee, C.W.; Chiang, Y.C.; Lee, M.H.; How, J.M.; Wu, C.M.; Huang, C.L.; Lee, I.T. Resveratrol inhibits urban particulate matter-induced COX-2/PGE2 release in human fibroblast-like synoviocytes via the inhibition of activation of NADPH oxidase/ROS/NF-kappaB. Int. J. Biochem. Cell Biol. 2017, 88, 113–123. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Baltimore, D. microRNA regulation of inflammatory responses. Annu. Rev. Immunol. 2012, 30, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Nejad, C.; Stunden, H.J.; Gantier, M.P. A guide to miRNAs in inflammation and innate immune responses. FEBS J. 2018, 285, 3695–3716. [Google Scholar] [CrossRef]
- Mousavi, M.J.; Jamshidi, A.; Chopra, A.; Aslani, S.; Akhlaghi, M.; Mahmoudi, M. Implications of the noncoding RNAs in rheumatoid arthritis pathogenesis. J. Cell Physiol. 2018, 234, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Huo, R.; Xiao, L.; Zhu, X.; Xie, J.; Sun, S.; He, Y.; Zhang, J.; Sun, Y.; Zhou, Z.; et al. A novel p53/microRNA-22/Cyr61 axis in synovial cells regulates inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Chen, H.; Li, Y.; Zhong, H.; Sun, W.; Wang, J.; Zhang, T.; Ma, J.; Yan, S.; Zhang, J.; et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann. Rheum. Dis. 2018, 77, 1644–1652. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Mao, X.; Guo, Q.; Li, W.; Wang, X.; Li, G.; Jiang, Q.; Lin, N. A Novel Circulating miRNA-Based Model Predicts the Response to Tripterysium Glycosides Tablets: Moving Toward Model-Based Precision Medicine in Rheumatoid Arthritis. Front. Pharmacol. 2018, 9, 378. [Google Scholar] [CrossRef] [Green Version]
- Kolarz, B.; Majdan, M. Epigenetic aspects of rheumatoid arthritis: Contribution of non-coding RNAs. Semin. Arthritis Rheum. 2017, 46, 724–731. [Google Scholar] [CrossRef]
- Rider, C.F.; Yamamoto, M.; Gunther, O.P.; Hirota, J.A.; Singh, A.; Tebbutt, S.J.; Carlsten, C. Controlled diesel exhaust and allergen coexposure modulates microRNA and gene expression in humans: Effects on inflammatory lung markers. J. Allergy Clin. Immunol. 2016, 138, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Singh, A.; Sava, F.; Pui, M.; Tebbutt, S.J.; Carlsten, C. MicroRNA expression in response to controlled exposure to diesel exhaust: Attenuation by the antioxidant N-acetylcysteine in a randomized crossover study. Environ. Health Perspect 2013, 121, 670–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodosthenous, R.S.; Coull, B.A.; Lu, Q.; Vokonas, P.S.; Schwartz, J.D.; Baccarelli, A.A. Ambient particulate matter and microRNAs in extracellular vesicles: A pilot study of older individuals. Part. Fibre Toxicol. 2016, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Cui, B.; Hao, X. MicroRNA22 alleviates inflammation in ischemic stroke via p38 MAPK pathways. Mol. Med. Rep. 2019, 20, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, Q.; Jin, X.; Zou, X.; Wang, Y.; Hao, D.; Fu, F.; Jiao, W.; Zhang, C.; Lin, H.; et al. Dysifragilone A inhibits LPSinduced RAW264.7 macrophage activation by blocking the p38 MAPK signaling pathway. Mol. Med. Rep. 2018, 17, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Zeid, A.; Saad, M.; Soliman, E. MicroRNA 146a expression in rheumatoid arthritis: Association with tumor necrosis factor-alpha and disease activity. Genet. Test. Mol. Biomark. 2011, 15, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Ceribelli, A.; Nahid, M.A.; Satoh, M.; Chan, E.K. MicroRNAs in rheumatoid arthritis. FEBS Lett. 2011, 585, 3667–3674. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.H.; Hsu, C.C.; Muo, C.H.; Hsu, C.Y.; Liu, H.C.; Kao, C.H.; Chen, C.Y.; Chang, M.Y.; Hsu, Y.C. Air pollution exposure increases the risk of rheumatoid arthritis: A longitudinal and nationwide study. Environ. Int. 2016, 94, 495–499. [Google Scholar] [CrossRef]
- Anderson, R.; Meyer, P.W.; Ally, M.M.; Tikly, M. Smoking and Air Pollution as Pro-Inflammatory Triggers for the Development of Rheumatoid Arthritis. Nicotine Tob. Res. 2016, 18, 1556–1565. [Google Scholar] [CrossRef]
- Doody, K.M.; Bottini, N.; Firestein, G.S. Epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes. Epigenomics 2017, 9, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Trenkmann, M.; Brock, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Huber, L.C. Tumor necrosis factor alpha-induced microRNA-18a activates rheumatoid arthritis synovial fibroblasts through a feedback loop in NF-kappaB signaling. Arthritis Rheum. 2013, 65, 916–927. [Google Scholar] [CrossRef]
- Philippe, L.; Alsaleh, G.; Pichot, A.; Ostermann, E.; Zuber, G.; Frisch, B.; Sibilia, J.; Pfeffer, S.; Bahram, S.; Wachsmann, D.; et al. MiR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann. Rheum. Dis. 2013, 72, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Nakamachi, Y.; Ohnuma, K.; Uto, K.; Noguchi, Y.; Saegusa, J.; Kawano, S. MicroRNA-124 inhibits the progression of adjuvant-induced arthritis in rats. Ann. Rheum. Dis. 2016, 75, 601–608. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Y.; Shen, H.; Li, H.; Long, L.; Hui, L.; Xu, W. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett. 2013, 587, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Li, Y.; Liu, D.; Zhang, R.; Zhang, J. miR-137 effects on gastric carcinogenesis are mediated by targeting Cox-2-activated PI3K/AKT signaling pathway. FEBS Lett. 2014, 588, 3274–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, H. miR-137 inhibits renal cell carcinoma growth in vitro and in vivo. Oncol. Lett. 2016, 12, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhang, F.; Guo, J. miR137 decreases proliferation, migration and invasion in rheumatoid arthritis fibroblastlike synoviocytes. Mol. Med. Rep. 2018, 17, 3312–3317. [Google Scholar] [CrossRef] [Green Version]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef]
- Rajaei, E.; Mowla, K.; Hayati, Q.; Ghorbani, A.; Dargahi-Malamir, M.; Hesam, S.; Zayeri, Z.D. Evaluating the relationship between serum level of interleukin-6 and rheumatoid arthritis severity and disease activity. Curr. Rheumatol. Rev. 2019. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, H.; Zhang, H.; Bai, J.; Zhang, Z. Interleukins 6/8 and cyclooxygenase-2 release and expressions are regulated by oxidative stress-JAK2/STAT3 signaling pathway in human bronchial epithelial cells exposed to particulate matter ≤ 2.5 μm. J. Appl. Toxicol. 2020. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.; Qiu, Q.; Xiao, Y.; Huang, M.; Shi, M.; Liang, L.; Yang, X.; Xu, H. Triptolide inhibits the migration and invasion of rheumatoid fibroblast-like synoviocytes by blocking the activation of the JNK MAPK pathway. Int. Immunopharmacol. 2016, 41, 8–16. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, S.W.; Baek, S.H.; Lee, C.W.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Suppression of PU.1-linked TLR4 expression by cilostazol with decrease of cytokine production in macrophages from patients with rheumatoid arthritis. Br. J. Pharmacol. 2013, 168, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khojah, H.M.; Ahmed, S.; Abdel-Rahman, M.S.; Hamza, A.B. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic. Biol. Med. 2016, 97, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Hyun, Y.M.; Kim, S.H.; Ko, M.K.; Park, C.O.; Hyun, J.W. Particulate matter induces inflammatory cytokine production via activation of NFkappaB by TLR5-NOX4-ROS signaling in human skin keratinocyte and mouse skin. Redox Biol. 2019, 21, 101080. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, M.; Li, Z.; Yue, J.; Xu, M.; Zhang, Y.; Yung, K.K.L.; Li, R. Fine particulate matter induces mitochondrial dysfunction and oxidative stress in human SH-SY5Y cells. Chemosphere 2019, 218, 577–588. [Google Scholar] [CrossRef]
- Rao, X.; Zhong, J.; Brook, R.D.; Rajagopalan, S. Effect of Particulate Matter Air Pollution on Cardiovascular Oxidative Stress Pathways. Antioxid. Redox Signal. 2018, 28, 797–818. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, M.-H.; Chi, M.-C.; Hsu, J.-F.; Lee, I.-T.; Lin, K.-M.; Fang, M.-L.; Lee, M.-H.; Lee, C.-W.; Liu, J.-F. Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis. Cells 2020, 9, 1378. https://doi.org/10.3390/cells9061378
Tsai M-H, Chi M-C, Hsu J-F, Lee I-T, Lin K-M, Fang M-L, Lee M-H, Lee C-W, Liu J-F. Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis. Cells. 2020; 9(6):1378. https://doi.org/10.3390/cells9061378
Chicago/Turabian StyleTsai, Ming-Horng, Miao-Ching Chi, Jen-Fu Hsu, I-Ta Lee, Ko-Ming Lin, Mei-Ling Fang, Ming-Hsueh Lee, Chiang-Wen Lee, and Ju-Fang Liu. 2020. "Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis" Cells 9, no. 6: 1378. https://doi.org/10.3390/cells9061378
APA StyleTsai, M. -H., Chi, M. -C., Hsu, J. -F., Lee, I. -T., Lin, K. -M., Fang, M. -L., Lee, M. -H., Lee, C. -W., & Liu, J. -F. (2020). Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis. Cells, 9(6), 1378. https://doi.org/10.3390/cells9061378