Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer

 ,
,  ,
,  , ,
, ,  and
and 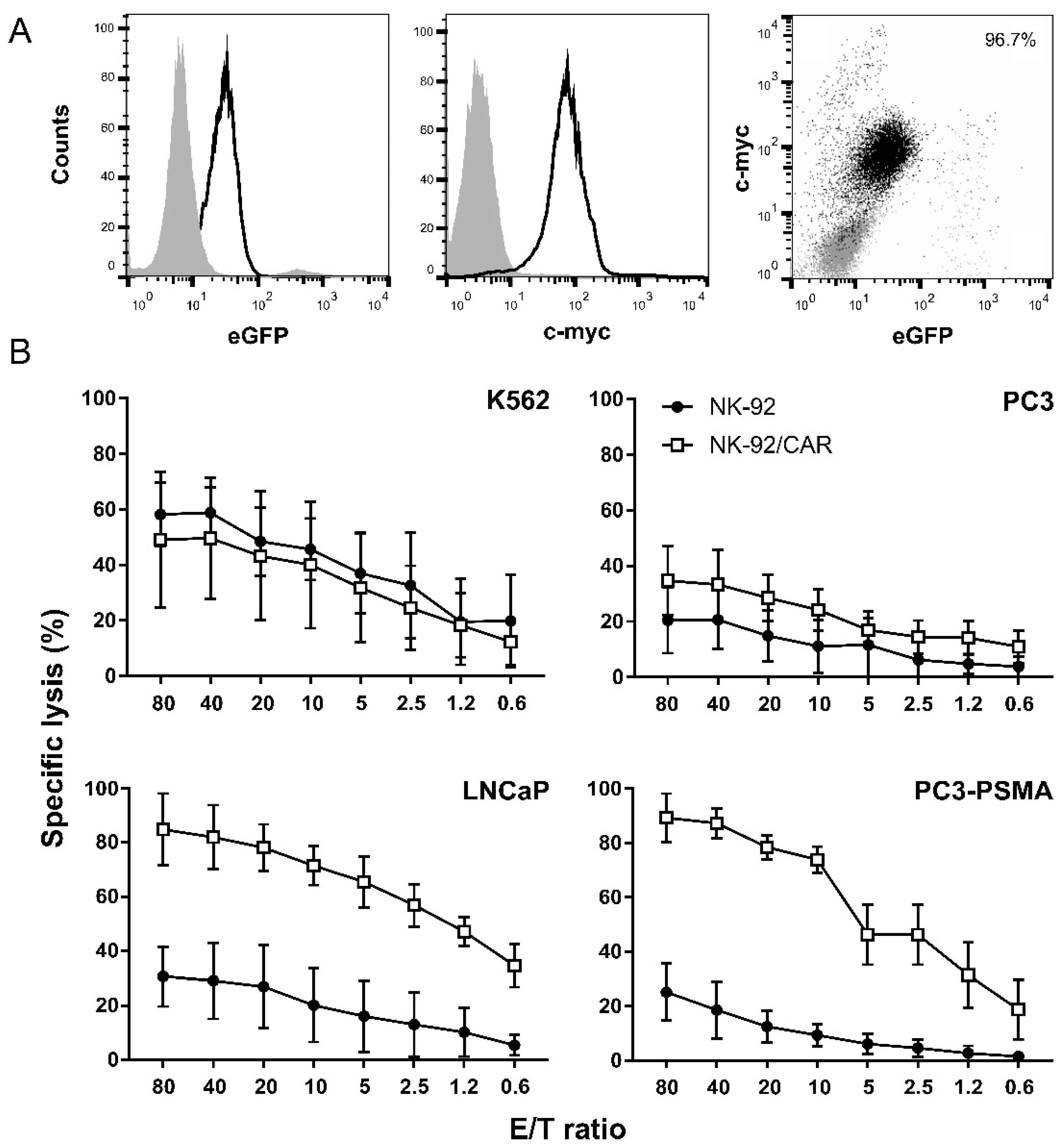{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Generation of CAR-Expressing NK-92 Cells
2.3. Irradiation of NK-92 Cells
2.4. Antibodies
2.5. Cytokine Release Assay
2.6. Degranulation Assay
2.7. Cytotoxicity Assay
2.8. In Vivo Studies
2.9. Statistics
3. Results
3.1. PSMA-Targeted NK-92/CAR Cells Acquire Antigen-Specific Cytotoxic Activity
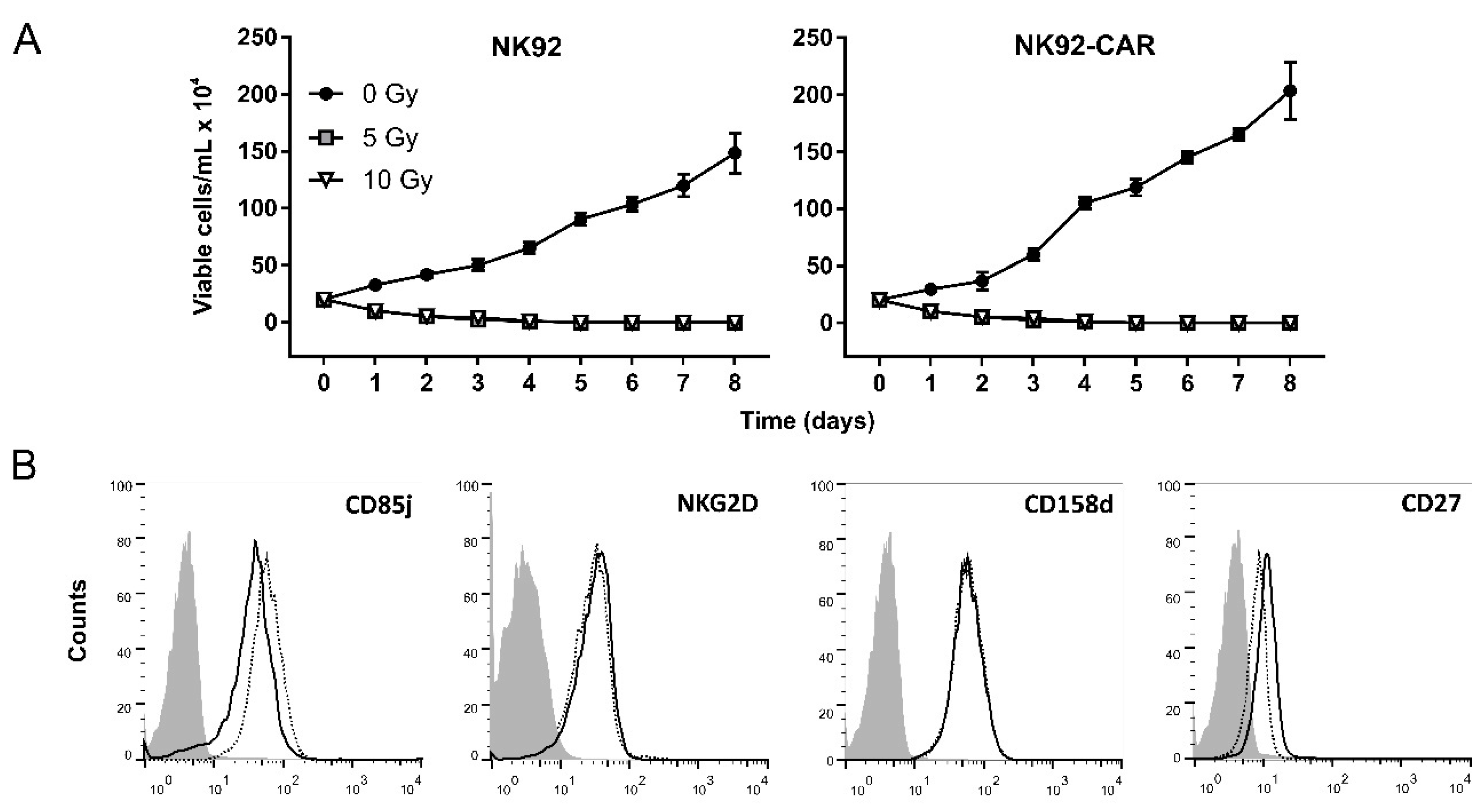
3.2. Irradiation Is a Safe Measure to Prevent NK-92/CAR Cell Proliferation
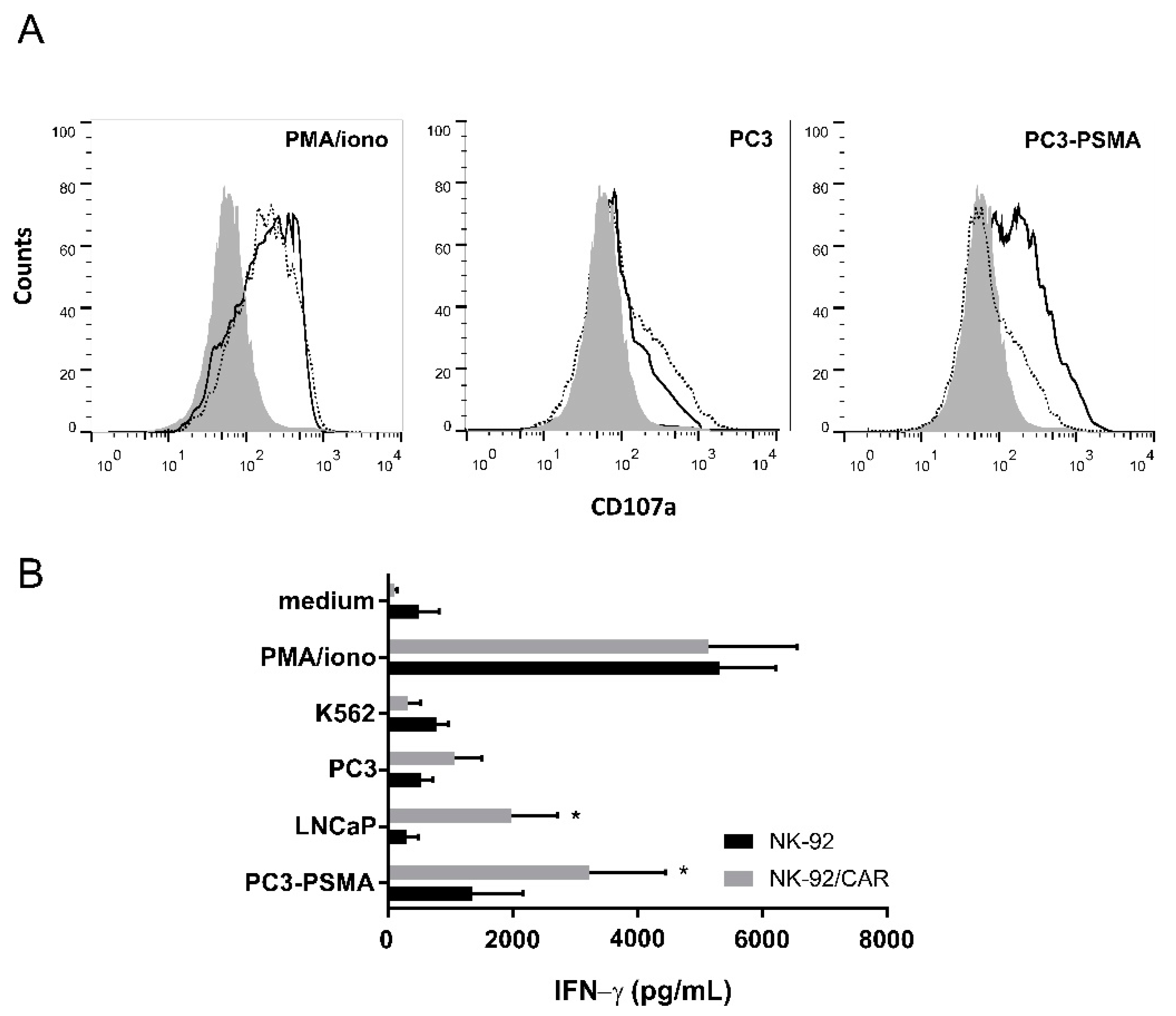
3.3. Irradiated NK-92/CAR Cells Undergo Degranulation and IFN-γ Release Upon Interaction with PSMA-Expressing Targets
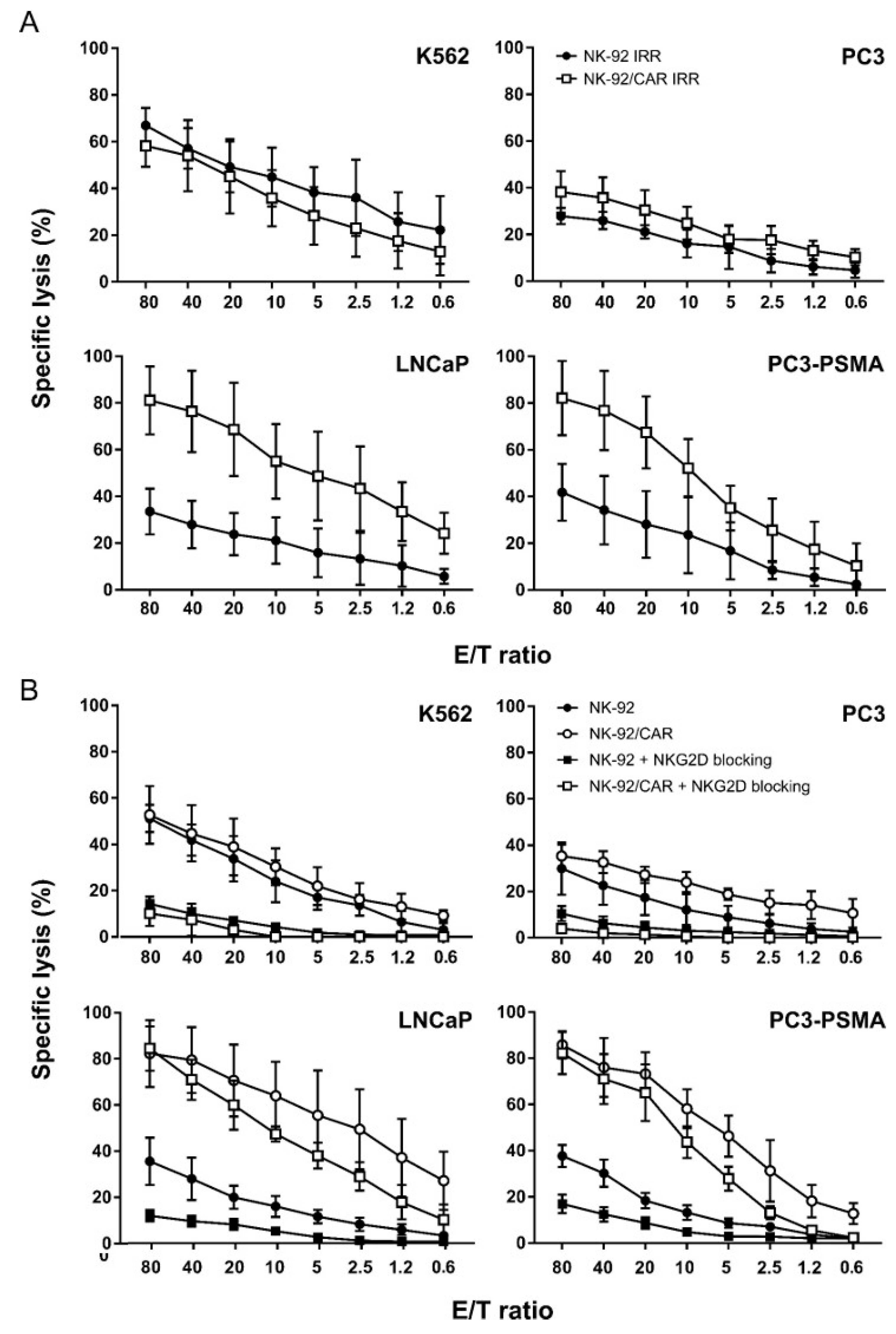
3.4. NK-92/CAR Cells Retain a High and Specific Target Killing Activity upon Irradiation
3.5. CAR-Mediated Cytotoxicity is Independent of NKG2D Activity in NK-92/CAR Cells
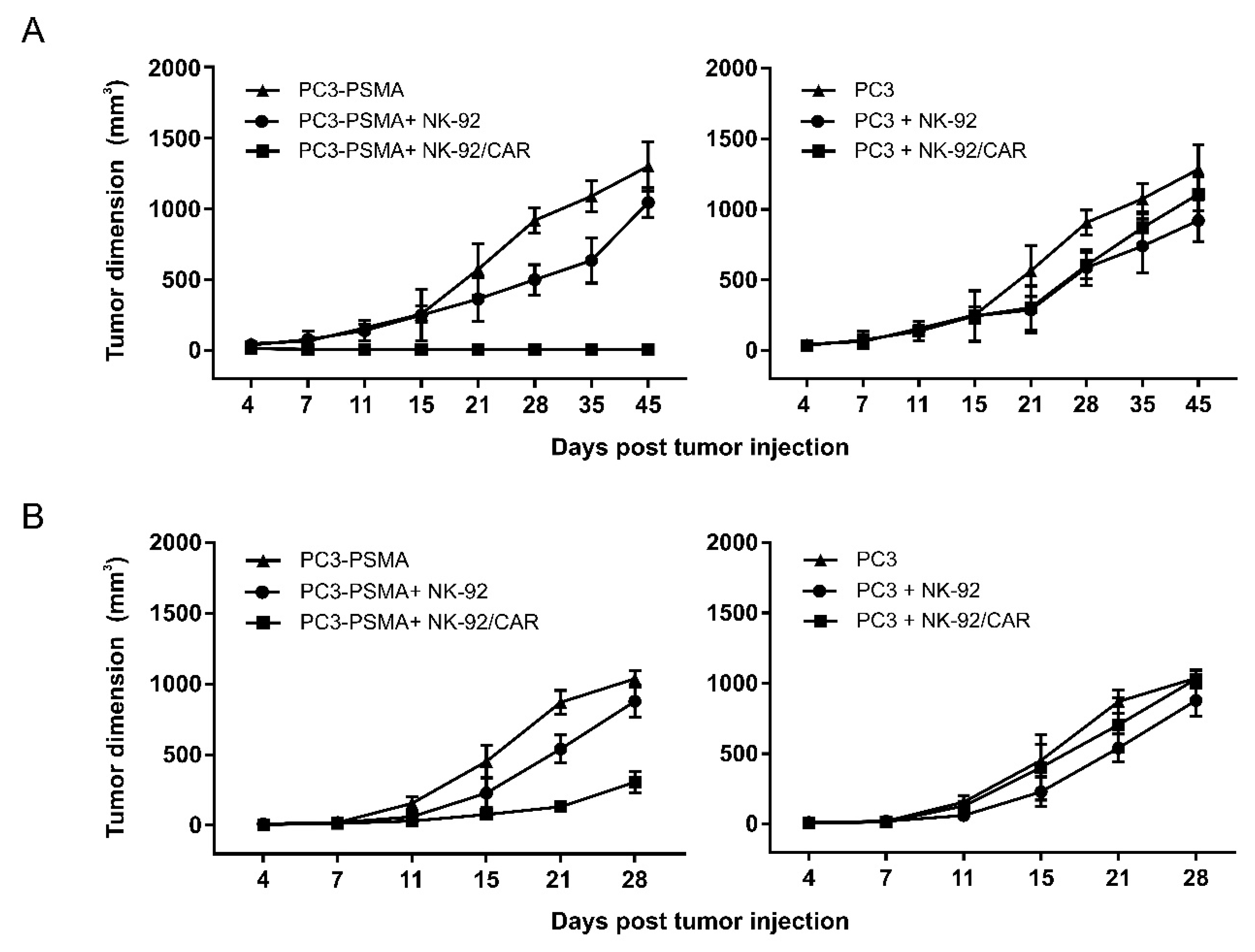
3.6. NK-92/CAR Cells Control the Growth of Locally Implanted Prostate Carcinoma
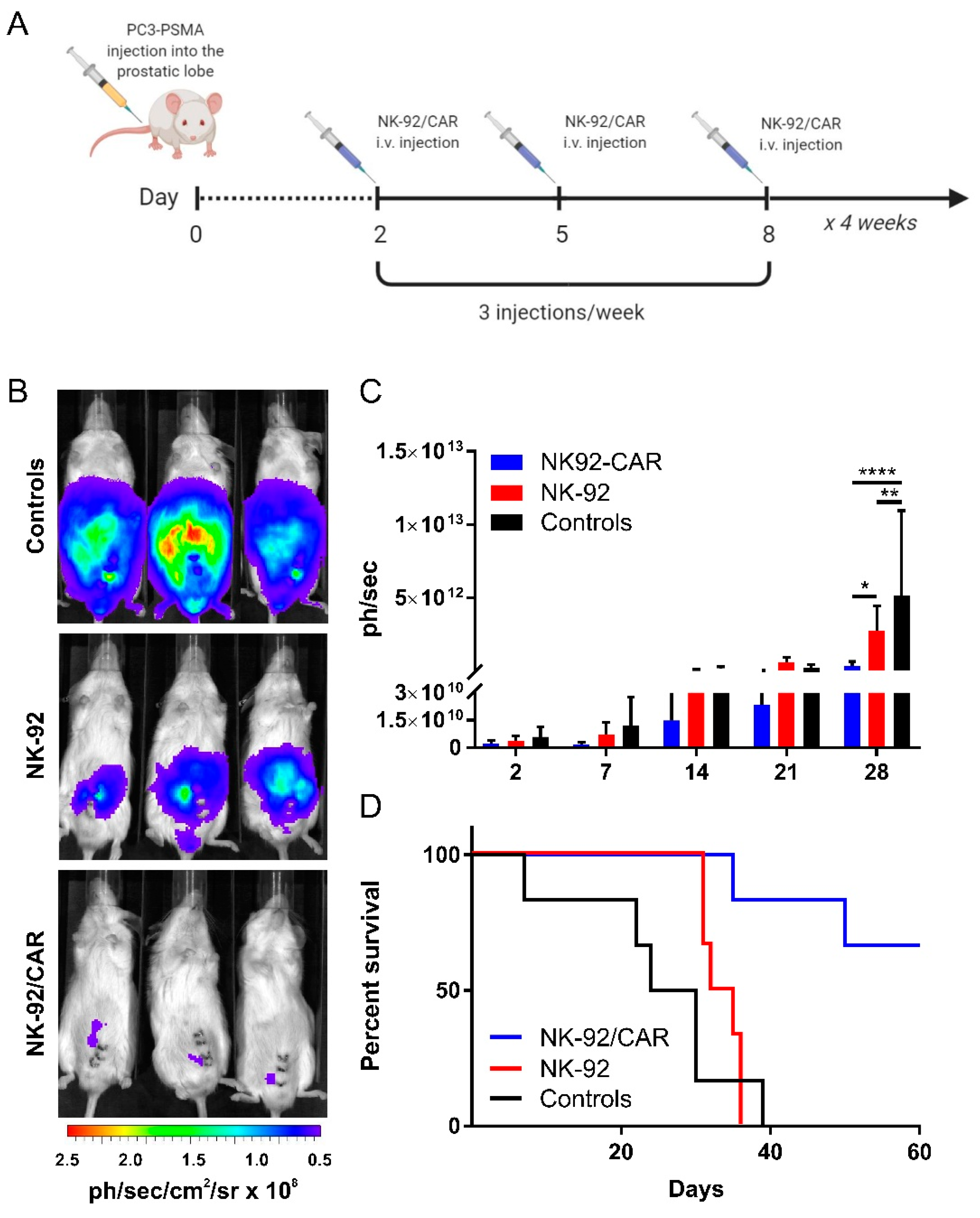
3.7. The Adoptive Transfer of NK-92/CAR Cells Is Therapeutic in an Orthotopic and Metastatic Prostate Tumor Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-Cell Therapy for Solid Tumors: Lessons Learned from Lymphoma Treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimasaki, N.; Coustan-Smith, E.; Kamiya, T.; Campana, D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy 2016, 18, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural killer cells for immunotherapy-Advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; den Hollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [Green Version]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Goding Sauer, A.; Ortiz, A.P.; Fedewa, S.A.; Pinheiro, P.S.; Tortolero-Luna, G.; Martinez-Tyson, D.; Jemal, A.; Siegel, R.L. Cancer Statistics for Hispanics/Latinos, 2018. CA Cancer J. Clin. 2018, 68, 425–445. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [Green Version]
- Ristau, B.T.; O’Keefe, D.S.; Bacich, D.J. The prostate-specific membrane antigen: Lessons and current clinical implications from 20 years of research. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Haberkorn, U.; Eder, M.; Kopka, K.; Babich, J.W.; Eisenhut, M. New strategies in prostate cancer: Prostate-specific membrane antigen (PSMA) ligands for diagnosis and therapy. Clin. Cancer Res. 2016, 22, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Zuccolotto, G.; Fracasso, G.; Merlo, A.; Montagner, I.M.; Rondina, M.; Bobisse, S.; Figini, M.; Cingarlini, S.; Colombatti, M.; Zanovello, P.; et al. PSMA-Specific CAR-Engineered T Cells Eradicate Disseminated Prostate Cancer in Preclinical Models. PLoS ONE 2014, 9, e109427. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Wang, X.; Klein, E.; Heston, W.D.W. Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res. 2005, 65, 727–731. [Google Scholar]
- Rosato, A.; Milan, G.; Collavo, D.; Zanovello, P. DNA-based vaccination against tumors expressing the P1A antigen. Methods 1999, 19, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-N.; Zhou, G.-Y.; Zhang, W.-L. NK-92 cell, another ideal carrier for chimeric antigen receptor. Immunotherapy 2017, 9, 753–765. [Google Scholar] [CrossRef]
- Ledford, H. Melanoma drug wins US approval. Nature 2011, 471, 561. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Simsek, M.; Basol Tekin, S.; Bilici, M. Immunological Agents Used in Cancer Treatment. Eurasian J. Med. 2019, 51, 90–94. [Google Scholar] [CrossRef]
- Piechocki, M.P.; Ho, Y.-S.; Pilon, S.; Wei, W.-Z. Human ErbB-2 (Her-2) Transgenic Mice: A Model System for Testing Her-2 Based Vaccines. J. Immunol. 2003, 171, 5787–5794. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef] [Green Version]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Lu, W.; Krause, D.; Van Etten, R.; Wels, W.; Klingemann, H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology 2013, 2, e26527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, R.; Müller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schönfeld, K.; et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell. Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef]
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.-G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. JNCI J. Natl. Cancer Inst. 2016, 108, 1–12. [Google Scholar] [CrossRef]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, B.; Sun, T.; Lin, L.; Hu, Y.; Deng, M.; Yang, J.; Liu, T.; Li, J.; Sun, S.; et al. Specific growth inhibition of ErbB2-expressing human breast cancer cells by genetically modified NK-92 cells. Oncol. Rep. 2014, 33, 95–102. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Martín-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Falcon, C.; Al-Obaidi, M.; Di Stasi, A. Exploiting Cell Death Pathways for Inducible Cell Elimination to Modulate Graft-versus-Host-Disease. Biomedicines 2017, 5, 30. [Google Scholar] [CrossRef]
- Thomis, D.C. A Fas-based suicide switch in human T cells for the treatment of graft-versus host disease. Blood 2001, 97, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Oliveira, G.; Stanghellini, M.T.L.; Vago, L.; Bondanza, A.; Peccatori, J. Improving the safety of cell therapy with the TK-suicide gene. Front. Pharmacol. 2015, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Gottschalk, S. CAR T Cells for Solid Tumors. Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montagner, I.M.; Penna, A.; Fracasso, G.; Carpanese, D.; Dalla Pietà, A.; Barbieri, V.; Zuccolotto, G.; Rosato, A. Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer. Cells 2020, 9, 1382. https://doi.org/10.3390/cells9061382
Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pietà A, Barbieri V, Zuccolotto G, Rosato A. Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer. Cells. 2020; 9(6):1382. https://doi.org/10.3390/cells9061382
Chicago/Turabian StyleMontagner, Isabella Monia, Alessandro Penna, Giulio Fracasso, Debora Carpanese, Anna Dalla Pietà, Vito Barbieri, Gaia Zuccolotto, and Antonio Rosato. 2020. "Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer" Cells 9, no. 6: 1382. https://doi.org/10.3390/cells9061382
APA StyleMontagner, I. M., Penna, A., Fracasso, G., Carpanese, D., Dalla Pietà, A., Barbieri, V., Zuccolotto, G., & Rosato, A. (2020). Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer. Cells, 9(6), 1382. https://doi.org/10.3390/cells9061382