Redirecting T Cells against Epstein–Barr Virus Infection and Associated Oncogenesis


Abstract
:1. Introduction of EBV and Its Oncogenesis
2. Immune Control of EBV
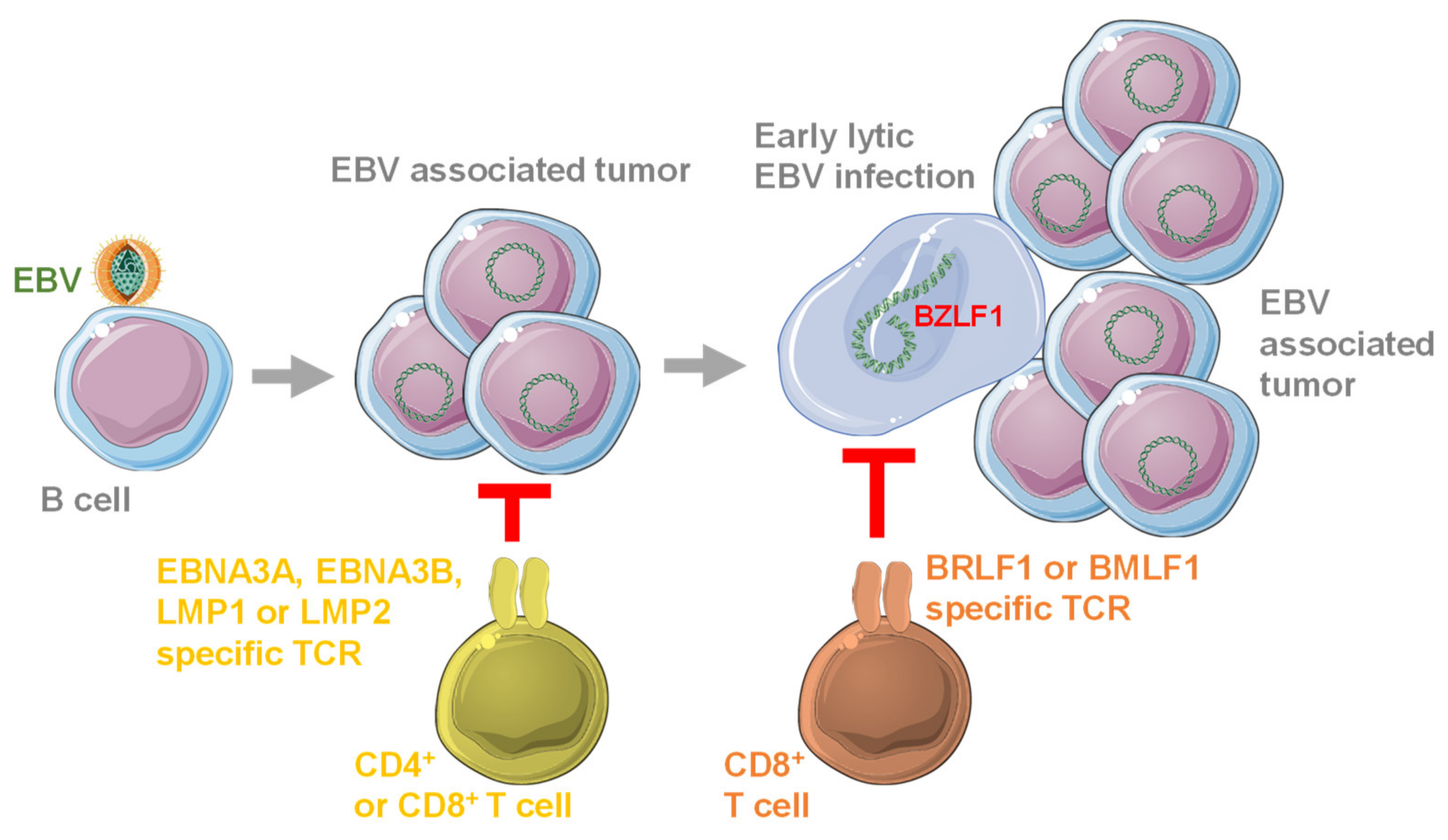
3. Antigen Specificity of Protective T Cell Responses against EBV
4. EBV Specific T Cell Receptors in Preclinical Studies
5. EBV Specific Chimeric Antigen Receptors in Preclinical Studies
6. Clinical Development of EBV Specific TCRs and B Cell Specific CARs against EBV Associated Malignancies
7. Conclusions and Outlook
Funding
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt’s lymphoma. J. Exp. Med. 1964, 121, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First days in the life of naive human B lymphocytes infected with Epstein-Barr virus. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [Green Version]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens-part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in the oncogenesis of the Epstein Barr virus. Nat. Rev. Micobiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef]
- Babcock, J.G.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, A.R.; Jankovic, M.; Eisenreich, T.; Difilippantonio, S.; Chen-Kiang, S.; Muramatsu, M.; Honjo, T.; Nussenzweig, A.; Nussenzweig, M.C. AID is required for c-myc/Igh chromosome translocations in vivo. Cell 2004, 118, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Murer, A.; McHugh, D.; Caduff, N.; Kalchschmidt, J.S.; Barros, M.H.; Zbinden, A.; Capaul, R.; Niedobitek, G.; Allday, M.J.; Chijioke, O.; et al. EBV persistence without its EBNA3A and 3C oncogenes in vivo. PLoS Pathog. 2018, 14, e1007039. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Kutok, J.L.; Wang, F. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 2006, 1, 375–404. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- Schober, T.; Framke, T.; Kreipe, H.; Schulz, T.F.; Grosshennig, A.; Hussein, K.; Baumann, U.; Pape, L.; Schubert, S.; Wingen, A.M.; et al. Characteristics of early and late PTLD development in pediatric solid organ transplant recipients. Transplantation 2013, 95, 240–246. [Google Scholar] [CrossRef]
- Totonchy, J.; Cesarman, E. Does persistent HIV replication explain continued lymphoma incidence in the era of effective antiretroviral therapy? Curr. Opin. Virol. 2016, 20, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.L.; Bollard, C.M. Cell therapies for hematological malignancies: Don’t forget non-gene-modified t cells! Blood Rev. 2018, 32, 203–224. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Rooney, C.M. Adoptive T-cell immunotherapy. Curr. Top Microbiol. Immunol. 2015, 391, 427–454. [Google Scholar] [PubMed] [Green Version]
- Smith, C.; Tsang, J.; Beagley, L.; Chua, D.; Lee, V.; Li, V.; Moss, D.J.; Coman, W.; Chan, K.H.; Nicholls, J.; et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. 2012, 72, 1116–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damania, B.; Münz, C. Immunodeficiencies that predispose to pathologies by human oncogenic gamma-herpesviruses. FEMS Microbiol. Rev. 2019, 43, 181–192. [Google Scholar] [CrossRef]
- Latour, S.; Fischer, A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef]
- Tangye, S.G.; Latour, S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood 2020, 135, 644–655. [Google Scholar] [CrossRef]
- McHugh, D.; Caduff, N.; Murer, A.; Engelmann, C.; Deng, Y.; Zdimerova, H.; Zens, K.; Chijioke, O.; Münz, C. Infection and immune control of human oncogenic gamma-herpesviruses in humanized mice. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180296. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Gurer, C.; Ploss, A.; Liu, Y.F.; Arrey, F.; Sashihara, J.; Koo, G.; Rice, C.M.; Young, J.W.; Chadburn, A.; et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 2009, 206, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajima, M.; Imadome, K.; Nakagawa, A.; Watanabe, S.; Terashima, K.; Nakamura, H.; Ito, M.; Shimizu, N.; Yamamoto, N.; Fujiwara, S. T cell-mediated control of Epstein-Barr virus infection in humanized mice. J. Infect. Dis. 2009, 200, 1611–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murer, A.; Ruhl, J.; Zbinden, A.; Capaul, R.; Hammerschmidt, W.; Chijioke, O.; Münz, C. MicroRNAs of Epstein-Barr virus attenuate T-cell-mediated immune control in vivo. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chijioke, O.; Muller, A.; Feederle, R.; Barros, M.H.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Landtwing, V.; Raykova, A.; Pezzino, G.; Beziat, V.; Marcenaro, E.; Graf, C.; Moretta, A.; Capaul, R.; Zbinden, A.; Ferlazzo, G.; et al. Cognate hla absence in trans diminishes human nk cell education. J. Clin. Investig. 2016, 126, 3772–3782. [Google Scholar] [CrossRef] [Green Version]
- Yuling, H.; Ruijing, X.; Li, L.; Xiang, J.; Rui, Z.; Yujuan, W.; Lijun, Z.; Chunxian, D.; Xinti, T.; Wei, X.; et al. EBV-induced human CD8+ NKT cells suppress tumorigenesis by ebv-associated malignancies. Cancer Res. 2009, 69, 7935–7944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Romero-Masters, J.C.; Hudson, A.W.; Gendron-Fitzpatrick, A.; Kenney, S.C.; Gumperz, J.E. Adoptively transferred Vgamma9Vdelta2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight 2017, 2, e93179. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Liu, Y.; Zheng, J.; Liu, M.; Lv, A.; Gao, Y.; Hu, H.; Lam, K.T.; Chan, G.C.; Yang, Y.; et al. Targeted activation of human Vgamma9Vdelta2-T cells controls Epstein-Barr virus-induced B cell lymphoproliferative disease. Cancer Cell 2014, 26, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Caduff, N.; McHugh, D.; Murer, A.; Ramer, P.; Raykova, A.; Landtwing, V.; Rieble, L.; Keller, C.W.; Prummer, M.; Hoffmann, L.; et al. Immunosuppressive FK506 treatment leads to more frequent EBV-associated lymphoproliferative disease in humanized mice. PLoS Pathog. 2020, 16, e1008477. [Google Scholar] [CrossRef] [Green Version]
- Chijioke, O.; Marcenaro, E.; Moretta, A.; Capaul, R.; Münz, C. The SAP-dependent 2B4 receptor mediates CD8+ T cell dependent immune control of Epstein Barr virus infection in mice with reconstituted human immune system components. J. Infect. Dis. 2015, 212, 803–807. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, B.; Deng, Y.; Holler, A.; Nunez, N.; Azzi, T.; Vanoaica, L.D.; Müller, A.; Zdimerova, H.; Antsiferova, O.; Zbinden, A.; et al. CD8+ T cells retain protective functions despite sustained inhibitory receptor expression during Epstein-Barr virus infection in vivo. PLoS Pathog. 2019, 15, e1007748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, Y.A.; Chen, Y.; Ong, H.S.; Wu, D.; Man, K.; Deleage, C.; Minnich, M.; Meckiff, B.J.; Wei, Y.; Hou, Z.; et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 2016, 17, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Danisch, S.; Slabik, C.; Cornelius, A.; Albanese, M.; Tagawa, T.; Chen, Y.A.; Kronke, N.; Eiz-Vesper, B.; Lienenklaus, S.; Bleich, A.; et al. Spatiotemporally skewed activation of programmed cell death receptor 1-positive T cells after Epstein-Barr virus infection and tumor development in long-term fully humanized mice. Am. J. Pathol. 2019, 189, 521–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujer, C.; Murer, A.; Muller, A.; Vanoaica, D.; Sutter, K.; Jacque, E.; Fournier, N.; Kalchschmidt, J.; Zbinden, A.; Capaul, R.; et al. Plasmacytoid dendritic cells respond to Epstein-Barr virus infection with a distinct type I interferon subtype profile. Blood Adv. 2019, 3, 1129–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Long, H.M.; Meckiff, B.J.; Taylor, G.S. The T-cell response to epstein-barr virus-new tricks from an old dog. Front. Immunol. 2019, 10, 2193. [Google Scholar] [CrossRef]
- Brooks, J.M.; Long, H.M.; Tierney, R.J.; Shannon-Lowe, C.; Leese, A.M.; Fitzpatrick, M.; Taylor, G.S.; Rickinson, A.B. Early T cell recognition of B cells following epstein-barr virus infection: Identifying potential targets for prophylactic vaccination. PLoS Pathog. 2016, 12, e1005549. [Google Scholar] [CrossRef]
- Blake, N.; Lee, S.; Redchenko, I.; Thomas, W.; Steven, N.; Leese, A.; Steigerwald-Mullen, P.; Kurilla, M.G.; Frappier, L.; Rickinson, A. Human CD8+ T cell responses to EBV EBNA1: Hla class I presentation of the (gly-ala)-containing protein requires exogenous processing. Immunity 1997, 7, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Münz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O’Donnell, M.; Steinman, R.M. Human CD4+ T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Long, H.M.; Haigh, T.A.; Gudgeon, N.H.; Leen, A.M.; Tsang, C.W.; Brooks, J.; Landais, E.; Houssaint, E.; Lee, S.P.; Rickinson, A.B.; et al. CD4+ T-cell responses to Epstein-Barr virus (EBV) latent-cycle antigens and the recognition of EBV-transformed lymphoblastoid cell lines. J. Virol. 2005, 79, 4896–4907. [Google Scholar] [CrossRef] [Green Version]
- Paludan, C.; Bickham, K.; Nikiforow, S.; Tsang, M.L.; Goodman, K.; Hanekom, W.A.; Fonteneau, J.F.; Stevanovic, S.; Münz, C. EBNA1 specific CD4+ Th1 cells kill Burkitt’s lymphoma cells. J. Immunol. 2002, 169, 1593–1603. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.K.P.; Hui, K.F.; Ning, R.J.; Xu, X.Q.; Chan, K.H.; Chiang, A.K.S. Emergence of CD4+ and CD8+ polyfunctional T cell responses against immunodominant lytic and latent EBV antigens in children with primary EBV infection. Front. Microbiol. 2018, 9, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meckiff, B.J.; Ladell, K.; McLaren, J.E.; Ryan, G.B.; Leese, A.M.; James, E.A.; Price, D.A.; Long, H.M. Primary EBV infection induces an acute wave of activated antigen-specific cytotoxic CD4+ Tt cells. J. Immunol. 2019, 203, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of Epstein-Barr virus (EBV) nuclear antigen 1-specific T cells as treatment for ebv reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013, 31, 39–48. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.G.; Kim, N.; Sohn, H.J.; Lee, S.K.; Oh, S.T.; Lee, H.J.; Cho, H.I.; Yim, H.W.; Jung, S.E.; Park, G.; et al. Long-term outcome of extranodal nk/t cell lymphoma patients treated with postremission therapy using EBV LMP1 and LMP2a-specific CTLs. Mol. Ther. 2015, 23, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zyl, D.G.; Tsai, M.H.; Shumilov, A.; Schneidt, V.; Poirey, R.; Schlehe, B.; Fluhr, H.; Mautner, J.; Delecluse, H.J. Immunogenic particles with a broad antigenic spectrum stimulate cytolytic T cells and offer increased protection against EBV infection ex vivo and in mice. PLoS Pathog. 2018, 14, e1007464. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, J.; Citterio, C.; Engelmann, C.; Haigh, T.A.; Dzionek, A.; Dreyer, J.H.; Khanna, R.; Taylor, G.S.; Wilson, J.B.; Leung, C.S.; et al. Heterologous prime-boost vaccination protects from EBV antigen expressing lymphomas. J. Clin. Investig. 2019, 129, 2071–2087. [Google Scholar] [CrossRef]
- Callan, M.F.; Tan, L.; Annels, N.; Ogg, G.S.; Wilson, J.D.; O’Callaghan, C.A.; Steven, N.; McMichael, A.J.; Rickinson, A.B. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J. Exp. Med. 1998, 187, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/CHK2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landais, E.; Saulquin, X.; Scotet, E.; Trautmann, L.; Peyrat, M.A.; Yates, J.L.; Kwok, W.W.; Bonneville, M.; Houssaint, E. Direct killing of Epstein-Barr virus (EBV)-infected B cells by CD4 T cells directed against the EBV lytic protein BHRF1. Blood 2004, 103, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef]
- Antsiferova, O.; Müller, A.; Rämer, P.; Chijioke, O.; Chatterjee, B.; Raykova, A.; Planas, R.; Sospedra, M.; Shumilov, A.; Tsai, M.H.; et al. Adoptive transfer of EBV specific CD8+ t cell clones can transiently control EBV infection in humanized mice. PLoS Pathog. 2014, 10, e1004333. [Google Scholar] [CrossRef]
- Linnerbauer, S.; Behrends, U.; Adhikary, D.; Witter, K.; Bornkamm, G.W.; Mautner, J. Virus and autoantigen-specific CD4+ T cells are key effectors in a scid mouse model of EBV-associated post-transplant lymphoproliferative disorders. PLoS Pathog. 2014, 10, e1004068. [Google Scholar] [CrossRef] [Green Version]
- Orentas, R.J.; Roskopf, S.J.; Nolan, G.P.; Nishimura, M.I. Retroviral transduction of a T cell receptor specific for an Epstein-Barr virus-encoded peptide. Clin. Immunol. 2001, 98, 220–228. [Google Scholar] [CrossRef]
- Schaft, N.; Lankiewicz, B.; Drexhage, J.; Berrevoets, C.; Moss, D.J.; Levitsky, V.; Bonneville, M.; Lee, S.P.; McMichael, A.J.; Gratama, J.W.; et al. T cell re-targeting to ebv antigens following tcr gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNgamma production. Int. Immunol. 2006, 18, 591–601. [Google Scholar] [CrossRef]
- Kobayashi, E.; Mizukoshi, E.; Kishi, H.; Ozawa, T.; Hamana, H.; Nagai, T.; Nakagawa, H.; Jin, A.; Kaneko, S.; Muraguchi, A. A new cloning and expression system yields and validates tcrs from blood lymphocytes of patients with cancer within 10 days. Nat. Med. 2013, 19, 1542–1546. [Google Scholar] [CrossRef]
- Cho, H.I.; Kim, U.H.; Shin, A.R.; Won, J.N.; Lee, H.J.; Sohn, H.J.; Kim, T.G. A novel Epstein-Barr virus-latent membrane protein-1-specific T-cell receptor for TCR gene therapy. Br. J. Cancer 2018, 118, 534–545. [Google Scholar] [CrossRef] [Green Version]
- Subklewe, M.; Sebelin, K.; Block, A.; Meier, A.; Roukens, A.; Paludan, C.; Fonteneau, J.F.; Steinman, R.M.; Münz, C. Dendritic cells expand Epstein Barr virus specific CD8+ T cell responses more efficiently than ebv transformed B cells. Hum. Immunol. 2005, 66, 938–949. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Christian, R.E.; Brooks, J.M.; Shabanowitz, J.; Settlage, R.E.; Marto, J.A.; White, F.M.; Rickinson, A.B.; Hunt, D.F.; Engelhard, V.H. Immunodominance among EBV-derived epitopes restricted by HLA-B27 does not correlate with epitope abundance in EBV-transformed B-lymphoblastoid cell lines. J. Immunol. 2000, 164, 6120–6129. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Shao, Q.; Sun, H.; Mu, X.; Gao, Y.; Jiang, R.; Hou, J.; Yao, K.; Chen, Y.; Sun, B. Evaluation of Epstein-Barr virus latent membrane protein 2 specific T-cell receptors driven by T-cell specific promoters using lentiviral vector. Clin. Dev. Immunol. 2011, 2011, 716926. [Google Scholar] [CrossRef] [Green Version]
- Frumento, G.; Zheng, Y.; Aubert, G.; Raeiszadeh, M.; Lansdorp, P.M.; Moss, P.; Lee, S.P.; Chen, F.E. Cord blood T cells retain early differentiation phenotype suitable for immunotherapy after TCR gene transfer to confer EBV specificity. Am. J. Transplant. 2013, 13, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.A.; Gao, L.; Ahmadi, M.; Ghorashian, S.; Barros, R.D.; Pospori, C.; Holler, A.; Wright, G.; Thomas, S.; Topp, M.; et al. Human MHC class I-restricted high avidity CD4 T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology 2013, 2, e22590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Parsonage, G.; Zhuang, X.; Machado, L.R.; James, C.H.; Salman, A.; Searle, P.F.; Hui, E.P.; Chan, A.T.; Lee, S.P. Human leukocyte antigen (HLA) A*1101-restricted Epstein-Barr virus-specific T-cell receptor gene transfer to target nasopharyngeal carcinoma. Cancer Immunol. Res. 2015, 3, 1138–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoldo, B.; Rooney, C.M.; Di Stasi, A.; Abken, H.; Hombach, A.; Foster, A.E.; Zhang, L.; Heslop, H.E.; Brenner, M.K.; Dotti, G. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 2007, 110, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific t cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martinez-Paniagua, M.A.; Sanber, K.; et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia 2020. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Ruella, M.; June, C.H. Emerging cellular therapies for cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 car T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Rundles, C. Common variable immune deficiency: Case studies. Blood 2019, 134, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Tang, Q.; Mao, Y.; Huang, X.; Jia, L.; Zhu, J.; Feng, Z. CD137 co-stimulation improves the antitumor effect of LMP1-specific chimeric antigen receptor T cells in vitro and in vivo. Onco Targets Ther. 2019, 12, 9341–9350. [Google Scholar] [CrossRef]
- Tang, X.; Zhou, Y.; Li, W.; Tang, Q.; Chen, R.; Zhu, J.; Feng, Z. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J. Biomed. Res. 2014, 28, 468–475. [Google Scholar]
- Verweij, F.J.; Van Eijndhoven, M.A.; Hopmans, E.S.; Vendrig, T.; Wurdinger, T.; Cahir-McFarland, E.; Kieff, E.; Geerts, D.; Van der Kant, R.; Neefjes, J.; et al. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappaB activation. EMBO J. 2011, 30, 2115–2129. [Google Scholar] [CrossRef]
- Gottschalk, S.; Rooney, C.M.; Heslop, H.E. Post-transplant lymphoproliferative disorders. Annu. Rev. Med. 2005, 56, 29–44. [Google Scholar] [CrossRef]
- Korell, F.; Laier, S.; Sauer, S.; Veelken, K.; Hennemann, H.; Schubert, M.L.; Sauer, T.; Pavel, P.; Mueller-Tidow, C.; Dreger, P.; et al. Current challenges in providing good leukapheresis products for manufacturing of CAR-T cells for patients with relapsed/refractory NHL or ALL. Cells 2020, 9, 1225. [Google Scholar] [CrossRef]
- Thomas, S.; Klobuch, S.; Sommer, M.; Van Ewijk, R.; Theobald, M.; Meyer, R.G.; Herr, W. Human CD8+ memory and EBV-specific T cells show low alloreactivity in vitro and in CD34+ stem cell-engrafted NOD/scid/IL-2Rgammac null mice. Exp. Hematol. 2014, 42, 28–38. [Google Scholar] [CrossRef]
- Heslop, H.E.; Brenner, M.K.; Rooney, C.M. Donor T cells to treat ebv-associated lymphoma. N. Engl. J. Med. 1994, 331, 679–680. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study Name | Conditions | Interventions | Trial Phase | Locations |
|---|---|---|---|---|
| TCR transgenic T cells | ||||
| EBV-TCR-T Cells for EB Virus Infection After HSCT (NCT04156217) | PTLD | HLA-A2, -A11 and -A24 restricted TCRs expressed in allogeneic donor T cells | Phase 1 | Hebei (China) |
| EBV-TCR-T (YT-E001) for Patients With EBV-positive Recurrent or Metastatic NPC (NCT03648697) | NPC | HLA-A2, -A11 and -A24 restricted LMP1, LMP2 and EBNA1 specific TCRs expressed in autologous T cells | Phase 1/2 | Fujian (China) |
| Phase I Trial of LMP2 Antigen-specific TCR T-cell Therapy for Recurrent and Metastatic NPC Patients (NCT03925896) | NPC | HLA-A2, -A11 and -A24 restricted LMP2 specific TCRs expressed in autologous T cells | Phase 1 | Guangzhou (China) |
| CAR transgenic T cells | ||||
| In Vitro Expanded Allogeneic Epstein–Barr Virus Specific Cytotoxic T-Lymphocytes (EBV-CTLs) Genetically Targeted to the CD19 Antigen in B-cell Malignancies (NCT01430390) | ALL, Lymphoma | CD19 specific CAR expressed in allogeneic EBV specific T cells | Phase 1 | New York (USA) |
| EBV CTLs Expressing CD30 Chimeric Receptors For CD30+ Lymphoma (CARCD30) (NCT01192464) | HD, NHL | CD30 specific CAR expressed in autologous EBV specific T cells | Phase 1 | Houston (USA) |
| A New EBV Related Technologies of T Cells in Treating Malignant Tumors and Clinical Application (NCT02980315) | NPC | LMP1 specific CAR expressed in autologous T cells | Phase 1/2 | Nanjing (China) |
| T-cells or EBV Specific CTLs, Advanced B-Cell NHL and CLL (ATECRAB) (NCT00709033) | CLL, NHL | CD19 specific CAR expressed in autologous EBV specific T cells | Phase 1 | Houston (USA) |
| Allogeneic CD30.CAR-EBVSTs in Patients With Relapsed or Refractory CD30-Positive Lymphomas (NCT04288726) | ENKTL, HD. PTLD | CD30 specific CAR expressed in allogeneic EBV specific T cells | Phase 1 | Houston (USA) |
| CD19-CAR Immunotherapy for Childhood Acute Lymphoblastic Leukemia (ALL) (CD19TPALL) (NCT04288726) | ALL | CD19 specific CAR expressed in EBV specific allogeneic donor T cells | Phase 1/2 | Essen, Hannover, Frankfurt, Münster (Germany), Bristol, London (UK) |
| Phase I CD19/CD22 Chimeric Antigen Receptor (CAR) T Cells in Adults With Recurrent/Refractory B Cell Malignancies (NCT03233854) | DLBCL, ALL | CD19/CD22 specific CAR expressed in autologous T cells | Phase 1 | Palo Alto (USA) |
| CD19 CAR and PD-1 Knockout Engineered T Cells for CD19 Positive Malignant B-cell Derived Leukemia and Lymphoma (NCT03298828) | ALL, BL | CD19 specific CAR expressed on autologous PD-1 knock-out T cells | Phase 1 | Chongqing (China) |
| CARPALL: Immunotherapy With CD19 CAR T-cells for CD19+ Hematological Malignancies (NCT02443831) | ALL, BL | CD19 specific CAR expressed on autologous T cells | Phase 1 | London, Manchester (UK) |
| Genetically Modified T-cell Infusion Following Peripheral Blood Stem Cell Transplant in Treating Patients With Recurrent or High-Risk Non-Hodgkin Lymphoma (NCT01815749) | NHL | CD19 specific CAR expressed in autologous T cells | Phase 1 | Duarte (USA) |
| Cellular Immunotherapy Following Chemotherapy in Treating Patients With Recurrent Non-Hodgkin Lymphomas, Chronic Lymphocytic Leukemia or B-Cell Prolymphocytic Leukemia (NCT02153580) | NHL, CLL | CD19 specific CAR expressed in autologous T cells | Phase 1 | Duarte (USA) |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Münz, C. Redirecting T Cells against Epstein–Barr Virus Infection and Associated Oncogenesis. Cells 2020, 9, 1400. https://doi.org/10.3390/cells9061400
Münz C. Redirecting T Cells against Epstein–Barr Virus Infection and Associated Oncogenesis. Cells. 2020; 9(6):1400. https://doi.org/10.3390/cells9061400
Chicago/Turabian StyleMünz, Christian. 2020. "Redirecting T Cells against Epstein–Barr Virus Infection and Associated Oncogenesis" Cells 9, no. 6: 1400. https://doi.org/10.3390/cells9061400
APA StyleMünz, C. (2020). Redirecting T Cells against Epstein–Barr Virus Infection and Associated Oncogenesis. Cells, 9(6), 1400. https://doi.org/10.3390/cells9061400