TAZ Controls Helicobacter pylori-Induced Epithelial–Mesenchymal Transition and Cancer Stem Cell-Like Invasive and Tumorigenic Properties

 , , ,
, , ,  ,
,
Abstract
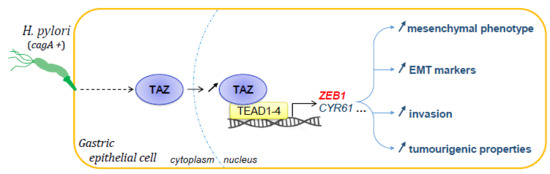
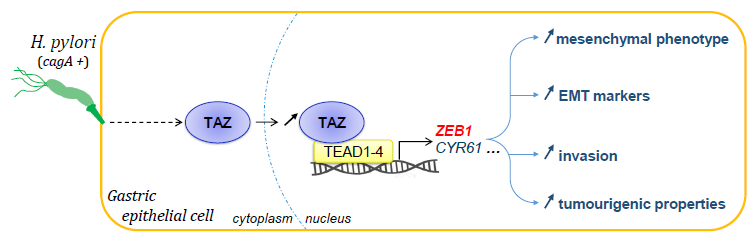
:
1. Introduction
2. Materials and Methods
2.1. Cell and Bacterial Culture and Co-Culture
2.2. Transfection of siRNA
2.3. TEAD-Luciferase Reporter Assay
2.4. RNA Extraction and RTqPCR
2.5. Western Blot
2.6. Immunofluorescence and Quantification of the Stainings
2.7. Tumorsphere Assay
2.8. Invasion Assay System
2.9. Immunohistochemistry
2.10. Ethic Statements on Human and Mouse Tissue Samples
2.11. Statistical Analysis
3. Results
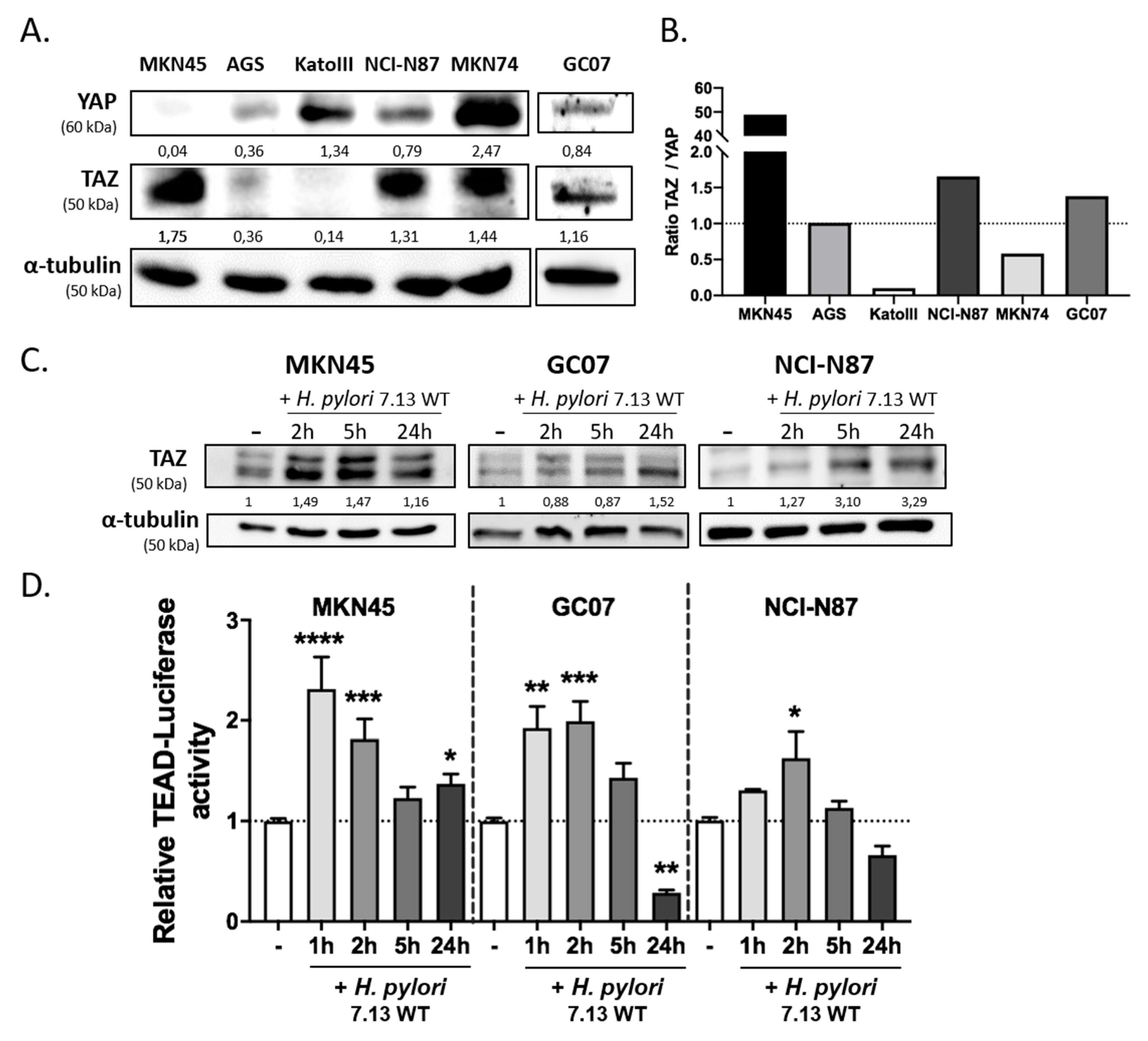
3.1. H. pylori Induced TAZ Overexpression and TAZ/TEAD-Dependent Transcriptional Activity
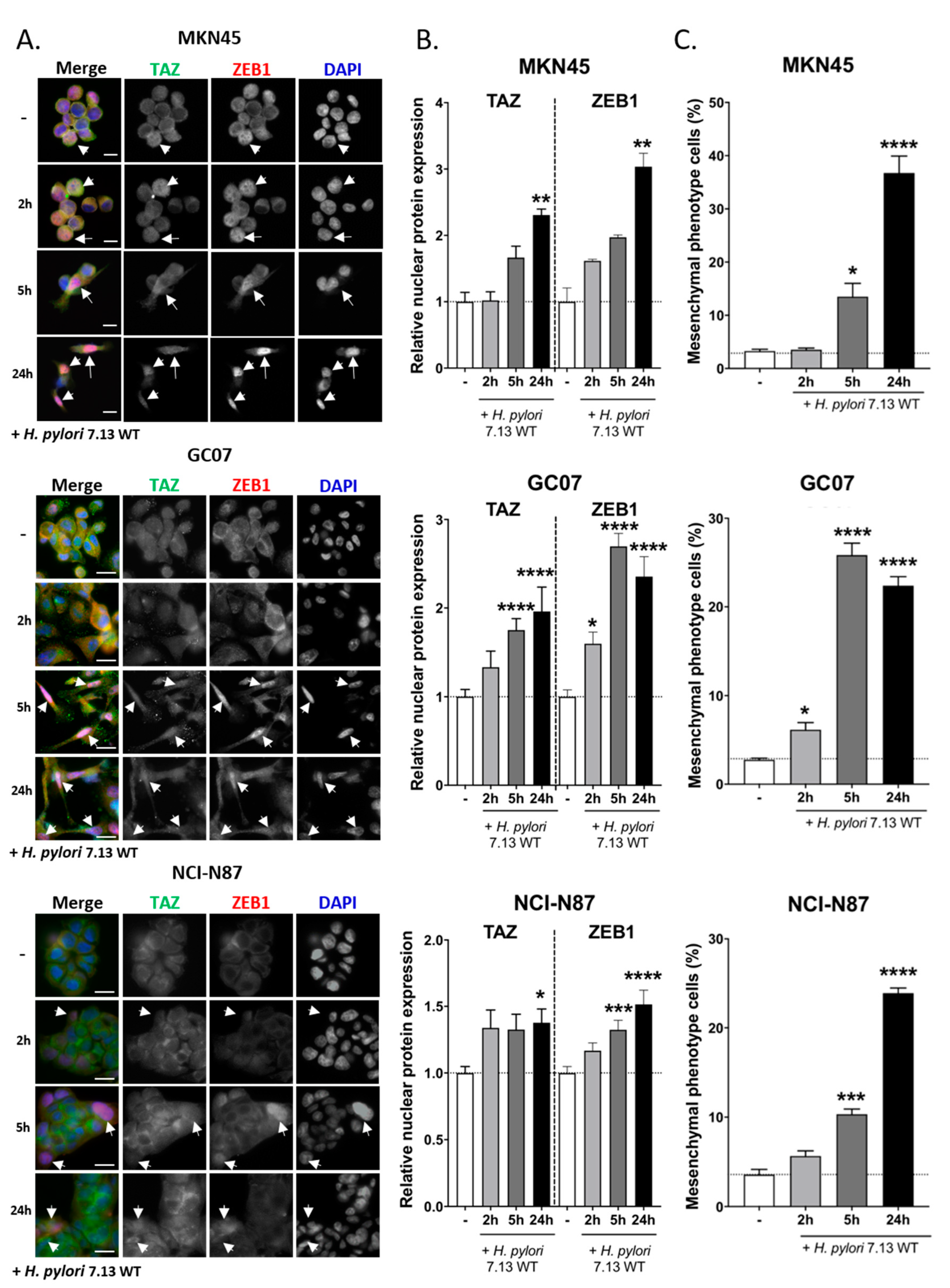
3.2. TAZ and ZEB1 Nuclear Co-Overexpression in H. pylori-Infected Gastric Epithelial Cells Was Associated with EMT and Was CagA-Dependent
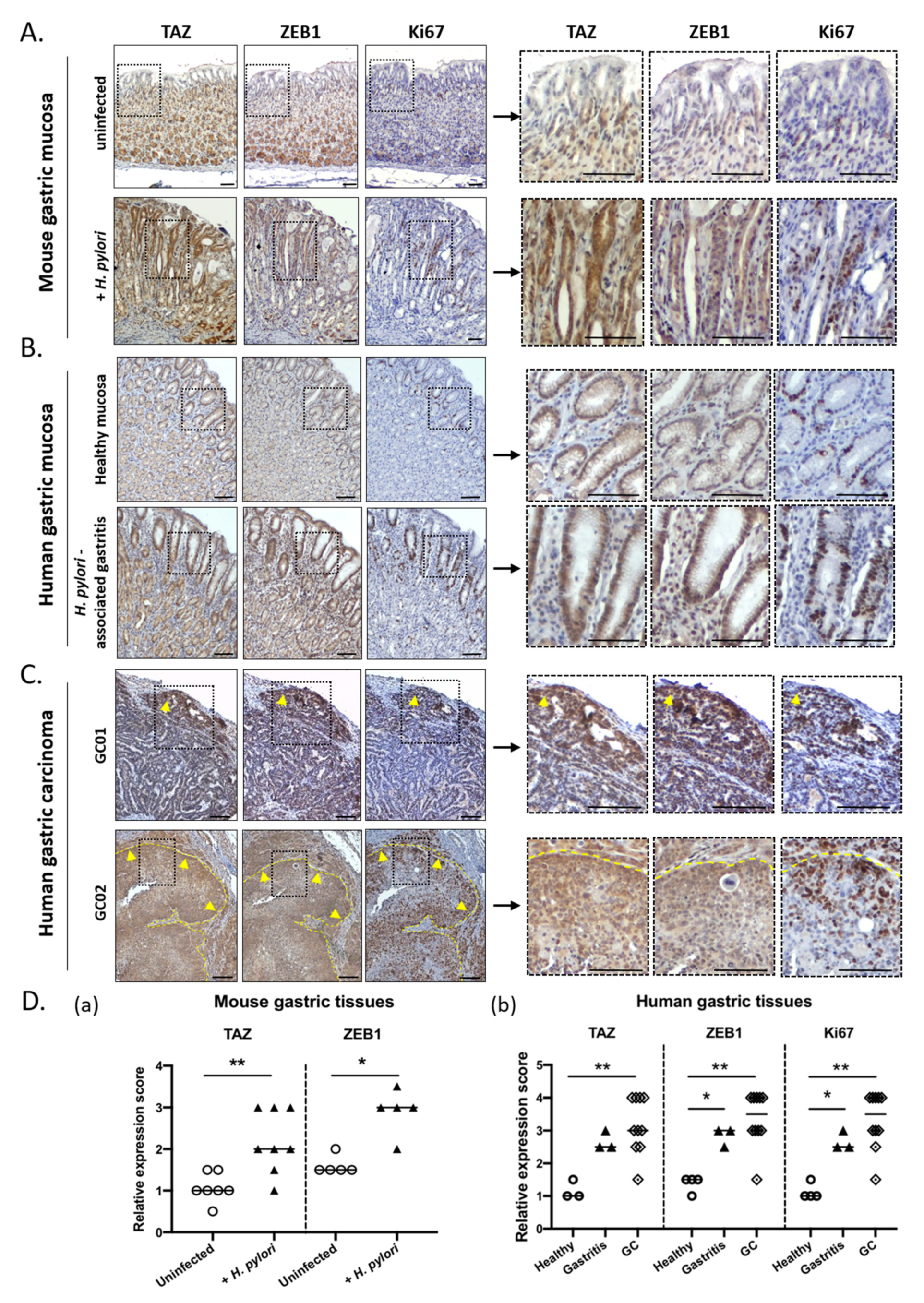
3.3. TAZ and ZEB1 Were Co-Upregulated in the Gastric Mucosa of H. pylori-Infected Patients and Mice and in Human Gastric Carcinoma Specimens
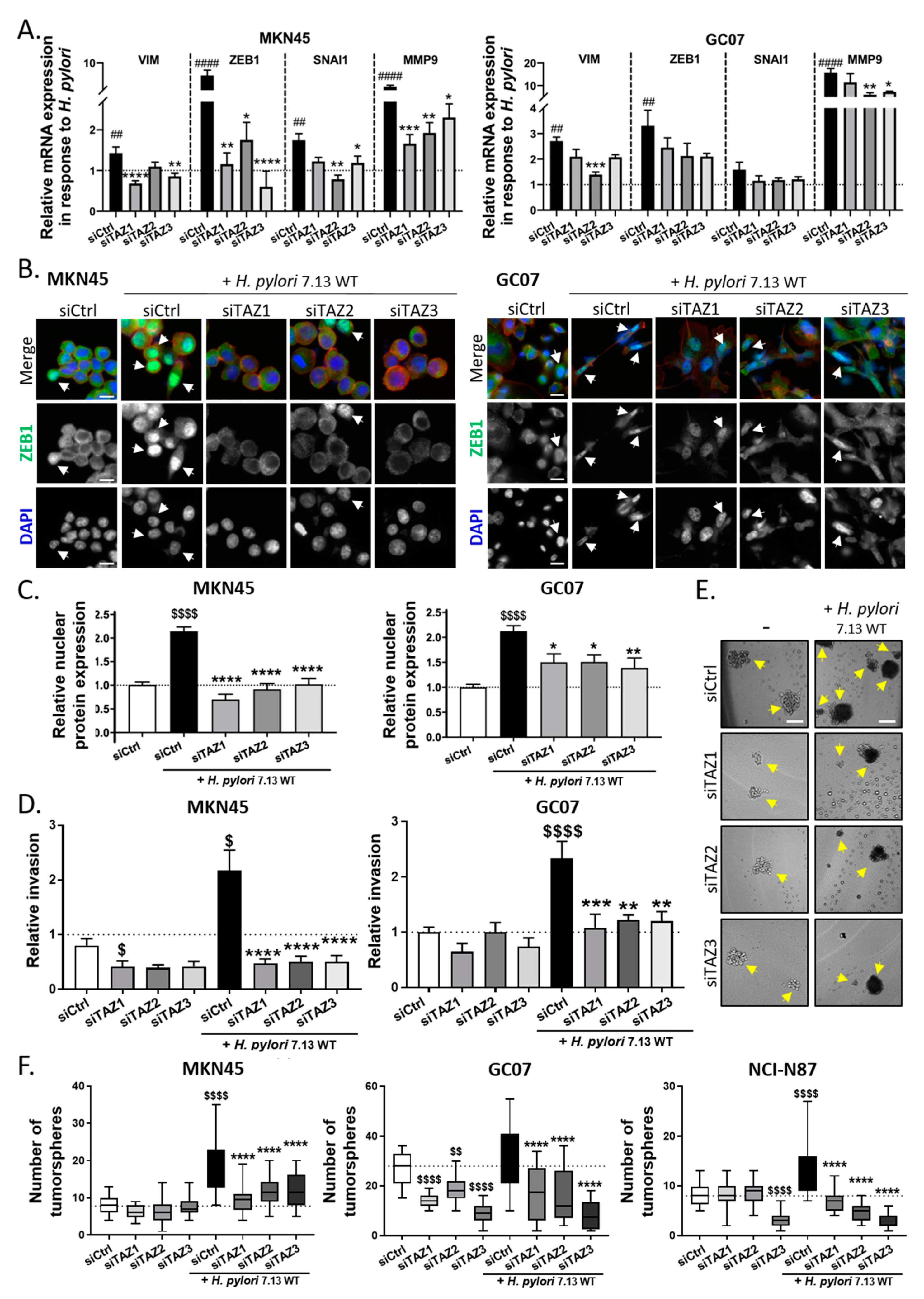
3.4. TAZ Conferred EMT Phenotype and CSC-like Properties via ZEB1 Upregulation in Response to H. pylori Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mégraud, F.; Bessède, E.; Varon, C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. 2015, 21, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, C.A.; Megraud, F.; Buissonniere, A.; Lujan Barroso, L.; Agudo, A.; Duell, E.J.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Palli, D.; Krogh, V.; et al. Helicobacter pylori infection assessed by ELISA and by immunoblot and noncardiac gastric cancer risk in a prospective study: The Eurgast-EPIC project. Ann. Oncol. 2012, 23, 1320–1324. [Google Scholar] [CrossRef]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar] [CrossRef]
- Backert, S.; Selbach, M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell. Microbiol. 2008, 10, 1573–1581. [Google Scholar] [CrossRef]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar] [CrossRef] [Green Version]
- Baud, J.; Varon, C.; Chabas, S.; Chambonnier, L.; Darfeuille, F.; Staedel, C. Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PLoS ONE 2013, 8, e60315. [Google Scholar] [CrossRef] [Green Version]
- Marques, M.S.; Melo, J.; Cavadas, B.; Mendes, N.; Pereira, L.; Carneiro, F.; Figueiredo, C.; Leite, M. Afadin downregulation by helicobacter pylori induces epithelial to mesenchymal transition in gastric cells. Front. Microbiol. 2018, 9, 2712. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Grabowska, A.M.; Clarke, P.A.; Whelband, E.; Robinson, K.; Argent, R.H.; Tobias, A.; Kumari, R.; Atherton, J.C.; Watson, S.A. Helicobacter pylori potentiates epithelial: Mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut 2010, 59, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Molina, S.; Acuña-Amador, L.; Dubus, P.; Staedel, C.; Chambonnier, L.; Buissonnière, A.; Sifré, E.; Giese, A.; Bénéjat, L.; et al. Deletion of IQGAP1 promotes Helicobacter pylori-induced gastric dysplasia in mice and acquisition of cancer stem cell properties in vitro. Oncotarget 2016, 7, 80688–80699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannée, G.; Mégraud, F.; Varon, C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 2014, 33, 4123–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, P.H.; Giraud, J.; Chambonnier, L.; Dubus, P.; Wittkop, L.; Belleannée, G.; Collet, D.; Soubeyran, I.; Evrard, S.; Rousseau, B.; et al. Characterization of biomarkers of tumorigenic and chemoresistant cancer stem cells in human gastric carcinoma. Clin. Cancer Res. 2017, 23, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Zhang, X.; Chu, C.; Cheung, W.L.; Ng, L.; Lam, S.; Chow, A.; Lau, T.; Chen, M.; Li, Y.; et al. Identification of CD44+ cancer stem cells in human gastric cancer. Hepatogastroenterology 2013, 60, 949–954. [Google Scholar] [CrossRef]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.W.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.K.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Saikawa, Y.; Ohashi, M.; Kumagai, K.; Kitajima, M.; Okano, H.; Matsuzaki, Y.; Kitagawa, Y. Tumor initiating potential of side population cells in human gastric cancer. Int. J. Oncol. 2009, 34, 1201–1207. [Google Scholar]
- Lau, W.M.; Teng, E.; Chong, H.S.; Lopez, K.A.P.; Tay, A.Y.L.; Salto-Tellez, M.; Shabbir, A.; So, J.B.Y.; Chan, S.L. CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res. 2014, 74, 2630–2641. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Bœuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; et al. All-trans retinoic acid targets gastric cancer stem cells and inhibits patient-derived gastric carcinoma tumor growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef]
- Courtois, S.; Durán, R.V.; Giraud, J.; Sifré, E.; Izotte, J.; Mégraud, F.; Lehours, P.; Varon, C.; Bessède, E. Metformin targets gastric cancer stem cells. Eur. J. Cancer 2017, 84, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo pathway in cancer: Aberrant regulation and therapeutic opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A nexus for hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Molina-Castro, S.E.; Tiffon, C.; Giraud, J.; Boeuf, H.; Sifre, E.; Giese, A.; Belleannée, G.; Lehours, P.; Bessède, E.; Mégraud, F.; et al. The hippo kinase LATS2 controls helicobacter pylori-induced epithelial-mesenchymal transition and intestinal metaplasia in gastric mucosa. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.; Tong, J.H.M.; Chan, A.W.H.; Lee, T.-L.; Lung, R.W.M.; Leung, P.P.S.; So, K.K.Y.; Wu, K.; Fan, D.; Yu, J.; et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin. Cancer Res. 2011, 17, 2130–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Cheong, J.-H.; Kim, H.; Noh, S.H.; Kim, H. Nuclear expression of Yes-associated protein 1 correlates with poor prognosis in intestinal type gastric cancer. Anticancer Res. 2012, 32, 3827–3834. [Google Scholar] [PubMed]
- Yu, L.; Gao, C.; Feng, B.; Wang, L.; Tian, X.; Wang, H.; Ma, D. Distinct prognostic values of YAP1 in gastric cancer. Tumour. Biol. 2017, 39, 1010428317695926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Zhu, Y.; Yuan, C.; Wang, D.; Zhang, W.; Qi, B.; Qiu, J.; Song, X.; Ye, J.; et al. The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol. Oncol. 2015, 9, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Cui, J.; Xia, T.; Jia, Z.; Wang, L.; Wei, W.; Zhu, A.; Gao, Y.; Xie, K.; Quan, M. Hippo transducer TAZ promotes epithelial mesenchymal transition and supports pancreatic cancer progression. Oncotarget 2015, 6, 35949–35963. [Google Scholar] [CrossRef] [Green Version]
- Yuen, H.-F.; McCrudden, C.M.; Huang, Y.-H.; Tham, J.M.; Zhang, X.; Zeng, Q.; Zhang, S.-D.; Hong, W. TAZ expression as a prognostic indicator in colorectal cancer. PLoS ONE 2013, 8, e54211. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.-H.; Hwang, E.S.; McManus, M.T.; Amsterdam, A.; Tian, Y.; Kalmukova, R.; Mueller, E.; Benjamin, T.; Spiegelman, B.M.; Sharp, P.A.; et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005, 309, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Wang, L.; Zhu, J.; Sun, A.; Yu, G.; Chen, M.; Huang, P.; Liu, H.; Shao, G.; Yang, W.; et al. The Hippo signaling effector WWTR1 is a metastatic biomarker of gastric cardia adenocarcinoma. Cancer Cell Int. 2019, 19, 74. [Google Scholar] [CrossRef]
- Yue, G.; Sun, X.; Gimenez-Capitan, A.; Shen, J.; Yu, L.; Teixido, C.; Guan, W.; Rosell, R.; Liu, B.; Wei, J. TAZ is highly expressed in gastric signet ring cell carcinoma. Biomed. Res. Int. 2014, 2014, 393064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.; Li, D.-S.; Chen, F.; Feng, J.-D.; Li, B.; Wang, T.-J. TAZ overexpression is associated with epithelial-mesenchymal transition in cisplatin-resistant gastric cancer cells. Int. J. Oncol. 2017, 51, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, J.; Molina-Castro, S.; Seeneevassen, L.; Sifré, E.; Izotte, J.; Tiffon, C.; Staedel, C.; Boeuf, H.; Fernandez, S.; Barthelemy, P.; et al. Verteporfin targeting YAP1/TAZ-TEAD transcriptional activity inhibits the tumorigenic properties of gastric cancer stem cells. Int. J. Cancer 2020, 146, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-X.; Li, X.-Y.; Zhang, Q.; Zhao, K.; Zhang, C.-P.; Xue, C.-H.; Yang, K.; Tian, Z.-B. Effects of the hippo signaling pathway in human gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5199–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, J.; Bouriez, D.; Seeneevassen, L.; Rousseau, B.; Sifré, E.; Giese, A.; Mégraud, F.; Lehours, P.; Dubus, P.; Gronnier, C.; et al. Orthotopic patient-derived xenografts of gastric cancer to decipher drugs effects on cancer stem cells and metastatic dissemination. Cancers (Basel) 2019, 11, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varon, C.; Dubus, P.; Mazurier, F.; Asencio, C.; Chambonnier, L.; Ferrand, J.; Giese, A.; Senant-Dugot, N.; Carlotti, M.; Mégraud, F. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology 2012, 142, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Odenbreit, S.; Püls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, L.; Liang, X.; Li, S.; Ma, L.; Zheng, L.; Li, T.; Yu, H.; Chan, H.; Chen, C.; et al. Helicobacter pylori-induced YAP1 nuclear translocation promotes gastric carcinogenesis by enhancing IL-1β expression. Cancer Med. 2019, 8, 3965–3980. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Feng, Y.; Hu, Y.; He, C.; Xie, C.; Ouyang, Y.; Artim, S.C.; Huang, D.; Zhu, Y.; Luo, Z.; et al. Helicobacter pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway. J. Exp. Clin. Cancer Res. 2018, 37, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Yu, G.; Shao, G.; Sun, A.; Chen, M.; Yang, W.; Lin, Q. CYR61 (CCN1) is a metastatic biomarker of gastric cardia adenocarcinoma. Oncotarget 2016, 7, 31067–31078. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Dubus, P.; Mégraud, F.; Varon, C. Helicobacter pylori infection and stem cells at the origin of gastric cancer. Oncogene 2015, 34, 2547–2555. [Google Scholar] [CrossRef] [PubMed]
- Weis, V.G.; Goldenring, J.R. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer 2009, 12, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Belair, C.; Baud, J.; Chabas, S.; Sharma, C.M.; Vogel, J.; Staedel, C.; Darfeuille, F. Helicobacter pylori interferes with an embryonic stem cell micro RNA cluster to block cell cycle progression. Silence 2011, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 2019, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Simeone, P.; Damato, M.; Maffia, M.; Lanuti, P.; Trerotola, M. The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J. Oncol. 2019, 2019, 1253727. [Google Scholar] [CrossRef]
- Oliveira, M.J.; Costa, A.M.; Costa, A.C.; Ferreira, R.M.; Sampaio, P.; Machado, J.C.; Seruca, R.; Mareel, M.; Figueiredo, C. CagA associates with c-Met, E-cadherin, and p120-catenin in a multiproteic complex that suppresses Helicobacter pylori-induced cell-invasive phenotype. J. Infect. Dis. 2009, 200, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Kanai, Y.; Oyama, T.; Yoshiura, K.; Shimoyama, Y.; Birchmeier, W.; Sugimura, T.; Hirohashi, S. E-cadherin gene mutations in human gastric carcinoma cell lines. Proc. Natl. Acad. Sci. USA 1994, 91, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Yamada, S.; Fuchs, B.C.; Fujii, T.; Nakayama, G.; Sugimoto, H.; Koike, M.; Fujiwara, M.; Tanabe, K.K.; Kodera, Y. Epithelial-to-mesenchymal transition predicts prognosis in clinical gastric cancer. J. Surg. Oncol. 2014, 109, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop–A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xin, Y.; Ye, F.; Wang, W.; Lu, Q.; Kaplan, H.J.; Dean, D.C. Taz-tead1 links cell-cell contact to zeb1 expression, proliferation, and dedifferentiation in retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3372–3378. [Google Scholar] [CrossRef]
- Vergara, D.; Ravaioli, S.; Fonzi, E.; Adamo, L.; Damato, M.; Bravaccini, S.; Pirini, F.; Gaballo, A.; Barbano, R.; Pasculli, B.; et al. Carbonic Anhydrase XII expression is modulated during epithelial mesenchymal transition and regulated through protein kinase C signaling. Int. J. Mol. Sci. 2020, 21, 715. [Google Scholar] [CrossRef] [Green Version]
- Posselt, G.; Wiesauer, M.; Chichirau, B.E.; Engler, D.; Krisch, L.M.; Gadermaier, G.; Briza, P.; Schneider, S.; Boccellato, F.; Meyer, T.F.; et al. Helicobacter pylori-controlled c-Abl localization promotes cell migration and limits apoptosis. Cell Commun. Signal 2019, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.J.; Jeong, H.; Han, K.H.; Kwon, H.M.; Hong, J.-H.; Hwang, E.S. TAZ suppresses NFAT5 activity through tyrosine phosphorylation. Mol. Cell. Biol. 2012, 32, 4925–4932. [Google Scholar] [CrossRef] [Green Version]
- Remue, E.; Meerschaert, K.; Oka, T.; Boucherie, C.; Vandekerckhove, J.; Sudol, M.; Gettemans, J. TAZ interacts with zonula occludens-1 and -2 proteins in a PDZ-1 dependent manner. FEBS Lett. 2010, 584, 4175–4180. [Google Scholar] [CrossRef]
- Nagy, T.A.; Frey, M.R.; Yan, F.; Israel, D.A.; Polk, D.B.; Peek, R.M. Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J. Infect. Dis. 2009, 199, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Nagy, T.A.; Wroblewski, L.E.; Wang, D.; Piazuelo, M.B.; Delgado, A.; Romero-Gallo, J.; Noto, J.; Israel, D.A.; Ogden, S.R.; Correa, P.; et al. β-Catenin and p120 mediate PPARδ-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology 2011, 141, 553–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal trans differentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Sequence Forward (5′-3′) | Sequence Reverse (5′-3′) |
|---|---|---|
| CYR61 | ATGAATTGATTGCAGTTGGAAA | TAAAGGGTTGTATAGGATGCGA |
| HPRT1 | TGGTCAGGCAGTATAATCCA | GGTCCTTTTCACCAGCAAGCT |
| MMP9 | Quantitect primer assay, cat. QT00040040 | |
| SNAI1 | ACAATGTCTGAAAAGGGACTGTGA | CAGACCAGAGCACCCCATT |
| TBP | GGGCATTATTTGTGCACTGAGA | GCCCAGATAGCAGCACGGT |
| VIM | GGATGCCCTTAAAGGAACCAA | CAACGGCAAAGTTCTCTTCCAT |
| ZEB1 | TCCCAACTTATGCCAGGCAC | CAGGAACCACATTTGTCATAGTCAC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiffon, C.; Giraud, J.; Molina-Castro, S.E.; Peru, S.; Seeneevassen, L.; Sifré, E.; Staedel, C.; Bessède, E.; Dubus, P.; Mégraud, F.; et al. TAZ Controls Helicobacter pylori-Induced Epithelial–Mesenchymal Transition and Cancer Stem Cell-Like Invasive and Tumorigenic Properties. Cells 2020, 9, 1462. https://doi.org/10.3390/cells9061462
Tiffon C, Giraud J, Molina-Castro SE, Peru S, Seeneevassen L, Sifré E, Staedel C, Bessède E, Dubus P, Mégraud F, et al. TAZ Controls Helicobacter pylori-Induced Epithelial–Mesenchymal Transition and Cancer Stem Cell-Like Invasive and Tumorigenic Properties. Cells. 2020; 9(6):1462. https://doi.org/10.3390/cells9061462
Chicago/Turabian StyleTiffon, Camille, Julie Giraud, Silvia Elena Molina-Castro, Sara Peru, Lornella Seeneevassen, Elodie Sifré, Cathy Staedel, Emilie Bessède, Pierre Dubus, Francis Mégraud, and et al. 2020. "TAZ Controls Helicobacter pylori-Induced Epithelial–Mesenchymal Transition and Cancer Stem Cell-Like Invasive and Tumorigenic Properties" Cells 9, no. 6: 1462. https://doi.org/10.3390/cells9061462
APA StyleTiffon, C., Giraud, J., Molina-Castro, S. E., Peru, S., Seeneevassen, L., Sifré, E., Staedel, C., Bessède, E., Dubus, P., Mégraud, F., Lehours, P., Martin, O. C. B., & Varon, C. (2020). TAZ Controls Helicobacter pylori-Induced Epithelial–Mesenchymal Transition and Cancer Stem Cell-Like Invasive and Tumorigenic Properties. Cells, 9(6), 1462. https://doi.org/10.3390/cells9061462