The S1P–S1PR Axis in Neurological Disorders—Insights into Current and Future Therapeutic Perspectives

 ,
, 

Abstract
:1. Introduction—S1P Metabolism and Signaling
1.1. De Novo Sphingolipid Synthesis and Signaling at the Endoplasmic Reticulum
1.2. Synthesis and Signaling of Sphingosine 1-Phosphate in Mitochondria and at the Plasma Membrane
1.3. Sphingosine 1-Phosphate Signaling in the Nucleus
1.4. Sphingosine 1-Phosphate in Autocrine and Paracrine Signaling
1.5. External Action of Sphingosine 1-Phosphate through S1PR
2. Implications of Sphingolipids in Neurological Disorders
2.1. The Sphingolipid Metabolism in Neurodegenerative Disorders
2.2. The Sphingolipid Metabolism in Neuroinflammatory Disorders
2.3. The Sphingolipid Metabolism in Cerebrovascular Diseases
3. Insights into Current and Future Therapeutic Perspectives
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Akt | RAC-alpha serine/threonine-protein kinase |
| ALS | Amyotrophic lateral sclerosis |
| aPC | Activated protein C |
| ApoM | Apolipoprotein M |
| APP | Amyloid precursor protein |
| Aβ | Amyloid-β-peptide |
| BBB | Blood brain barrier |
| CBAs | Conjugated bile acids |
| CBF | Cerebral blood flow |
| CCL4 | C-C motif chemokine 4-like |
| CCL5 | C-C motif chemokine 5-like |
| CCO | Cytochrome c oxidase |
| CCR7 | C-C chemokine receptor type 7 |
| CD | Cluster of differentiation molecule |
| CDase | Ceramidase |
| c-fos | Proto-oncogene c-Fos |
| cIAP2 | Cellular inhibitor of apoptosis 2 |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CXCL8 | C-X-C motif chemokine 8, IL-8 |
| CXCL10 | C-X-C motif chemokine 10 |
| DHHC5 | Palmitoyltransferase DHHC5 |
| DNA | Desoxyribonucleic acid |
| EAE | Experimental autoimmune encephalomyelitis |
| EC | Extracellular |
| ERK | Extracellular-regulated kinase |
| GBA | Glucocerebrosidase |
| GCS | Glucosylceramide synthase |
| Glc | Glucose |
| GlcCer | Glucosylceramidase |
| GRK2 | G protein-coupled receptor kinase 2 |
| Gαi/o | Inhibitory Gα subunit |
| Gαq | q subtype of Gα proteins |
| Gαs | Stimulatory Gα subunit |
| G12/13 | 12/13 subtype of Gα proteins |
| HD | Huntigton’s disease |
| HDAC1/2 | Histone deacetylases 1/2 |
| HDL | High-density lipoprotein |
| hTERT | Human telomerase reverse transcriptase |
| IC | Intracellular |
| ICAM1 | Intracellular adhesion molecule 1 |
| IFN-α | Interferon α |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| IL-17A | Interleukin-17A |
| IRF1 | Interferon regulatory factor 1 |
| IκB | Inhibitor of nuclear factor κ-B kinase |
| IκBα | NF-κB inhibitor α |
| LBD | Lewy body disorders |
| LDL | Low-density lipoprotein |
| LPP | Lipid phosphate phosphohydrolase |
| MCAO | Middle cerebral artery occlusion |
| MFSD2B | Major facilitator superfamily domain-containing protein 2B |
| MKRN1 | Makorin ring finger protein 1 |
| MMP-9 | Matrix metalloproteinase-9 |
| mRNA | Messenger ribonucleic acid |
| MS | Multiple slerosis |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NMO | Neuromyelitis optica |
| NVU | Neurovascular unit |
| ORMDL3 | ORM1-like protein 3 |
| OXPHOS | Oxidative phosphorylation |
| PalCoA | Palmitoyl-CoA |
| PD | Parkinson’s disease |
| PE | Phosphoethanolamine |
| PGC1β | PPARγ co-activator 1β |
| PHB2 | Prohibitin-2 |
| PKC | Protein kinase C |
| PKCζ | Atypical PKC subtype ζ |
| PKCι | Atypical PKC subtype ι |
| PLPP3 | Phospholipid phosphatase 3 |
| PM | Plasma membrane |
| PPARγ | Peroxisome proliferator activated receptor γ |
| PTX3 | Pentraxin-related protein 3 |
| p21 | Cyclin-dependent kinase inhibitor 1 |
| RACK | Receptor for activated C kinase proteins |
| RhoA | Transforming protein RhoA |
| RING | Really interesting new group |
| RIP1 | Receptor-interacting serine/threonine-protein kinase 1 |
| rt-PA | Recombinant-tissue plasminogen activator |
| SAH | Subarachnoid hemorrhage |
| sER | Smooth endoplasmic reticulum |
| SGPL1 | S1P lyase 1 |
| SGPP1/2 | Sphingosine 1-phosphate phosphohydrolase 1/2 |
| SLE | Systemic lupus erythematosus |
| SMase | Sphingomyelinase |
| SMS | Sphingomyelin synthase |
| SphK1 | Sphingosine kinase 1 |
| SphK2 | Sphingosine kinase 2 |
| SPNS2 | Spinster homolog 2 |
| SPT | Serine palmitoyltransferase |
| STAT3 | Signal transducer and activator of transcription 3 |
| S1P | Sphingosine 1-phosphate |
| S1PR | Sphingosine 1-phosphate receptor |
| S1P1–5 | S1PR subtype 1–5 |
| TAB3 | TGF-β-activated kinase 1 and MAP3K7-binding protein3 |
| TFH | Follicular TH cell |
| TGF-β | Transforming-growth-factor-β |
| TH-17 | TH-17 type cell |
| TNFRSF12A | Tumor necrosis factor receptor superfamily member 12A |
| TNF-α | Tumor necrosis factor-α |
| TRAF2 | TNF-α receptor-associated factor 2 |
| TRAF6 | TNF-α receptor-associated factor 6 |
| TREG | Regulatory T cell |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated, uridine 5′triphosphate-biotin nick end-labeling |
| UGT8A | UDP galactosyltransferase 8A |
| VEGFa | Vascular endothelial growth factor A |
| VLDL | Very low-density lipoprotein |
| WWP2 | NEDD4-like E3 ubiquitin ligase WWP2 |
References
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 114, 155–167. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of cermide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, D.K.; Weissman, J.S. Membranes in Balance: Mechanisms of Sphingolipid Homeostasis. Mol. Cell. 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. The ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar] [CrossRef] [Green Version]
- Linn, S.C.; Kim, H.S.; Keane, E.M.; Andras, L.M.; Wang, E.; Merrill, A.H., Jr. Regulation of de novo sphingolipid biosynthesis and the toxic consequences of its disruption. Biochem. Soc. Trans. 2001, 29, 831–835. [Google Scholar] [CrossRef]
- Xu, R.; Jin, J.; Hu, W.; Sun, W.; Bielawski, J.; Szulc, Z.; Taha, T.; Obeid, L.M.; Mao, C. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. FASEB J. 2006, 20, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Wu, B.X.; Mao, C.; Roddy, P.; El Bawab, S.; Hannun, Y.A. Identification of a novel amidase motif in neutral ceramidase. Biochem. J. 2006, 393, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinetti, A.; Loberto, N.; Chigorno, V.; Sonnino, S. Glycosphingolipid behaviour in complex membranes. Biochim. Biophys. Acta 2009, 1788, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Tafesse, F.G.; Ternes, P.; Holthuis, J.C.M. The multigenic sphingomyelin synthase family. J. Biol. Chem. 2006, 281, 29421–29425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakomori, S. Traveling for the glycosphingolipid path. Glycoconj. J. 2000, 17, 627–647. [Google Scholar] [CrossRef]
- Ichikawa, S.; Hirabayashi, Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. 1998, 8, 198–202. [Google Scholar] [CrossRef]
- Zhou, J.; Saba, J.D. Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem. Biophys. Res. Commun. 1998, 242, 502–507. [Google Scholar] [CrossRef]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase in immunity and cancer: Silencing the siren. Trends Mol. Med. 2007, 13, 210–217. [Google Scholar] [CrossRef]
- Nakahara, K.; Ohkuni, A.; Kitamura, T.; Abe, K.; Naganuma, T.; Ohno, J.; Zoeller, R.A.; Kihara, A. The Sjögren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell. 2012, 46, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Dobrosotskaya, I.Y.; Seegmiller, A.C.; Brown, M.S.; Goldstein, J.L.; Rawson, R.B. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 2002, 29, 879–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantalupo, A.; Zhang, Y.; Kothiya, M.; Galvani, S.; Obinata, H.; Bucci, M.; Giordano, F.J.; Jiang, X.-C.; Hla, T.; Di Lorenzo, A. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat. Med. 2015, 21, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, Y.; Cantalupo, A.; Azevedo, P.S.; Siragusa, M.; Bielawski, J.; Giordano, F.J.; Di Lorenzo, A. Endothelial Nogo-B regulates sphingolipid biosynthesis to promote pathological cardiac hypertrophy during chronic pressure overload. JCI Insight 2016, 1, e85484. [Google Scholar] [CrossRef] [Green Version]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lépine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, C.; Kihara, A.; Gokoh, M.; Igarashi, Y. Identification and characterization of a novel human sphingosine-1-phosphate phosphohydrolase, hSPP2. J. Biol. Chem. 2003, 278, 1268–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug. Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillard, B.K.; Clement, R.G.; Marcus, D.M. Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology 1998, 8, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, G.; Bassi, R.; Viani, P.; Riboni, L. Salvage pathways in glycosphingolipid metabolism. Biochimie 2003, 85, 423–437. [Google Scholar] [CrossRef]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Huwiler, A.; Kolter, T.; Pfeilschifter, J.; Sandhoff, K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim. Biophys. ACTA 2000, 1485, 63–99. [Google Scholar] [CrossRef]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.-W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a Sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS One 2012, 7, e38941. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest. 2012, 122, 1416–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef]
- Kajimoto, T.; Caliman, A.D.; Tobias, I.S.; Okada, T.; Pilo, C.A.; Van, A.-A. N.; McCammon, J.A.; Nakamura, S.-I.; Newton, A.C. Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter. Sci. Signal. 2019, 12, eaat6662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, a multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. The atypical protein kinase Cs: Functional specificity mediated by specific protein adapters. EMBO Rep. 2000, 1, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, L.; Dorf, M.E. PKC phosphorylation of TRAF2 mediates IKKα/β recruitment and K63-linked polyubiquitination. Mol. Cell. 2009, 33, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Etemadi, N.; Chopin, M.; Anderton, H.; Tanzer, M.C.; Rickard, J.A.; Abeysekera, W.; Hall, C.; Spall, S.; Wang, B.; Xiong, Y.; et al. TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of sphingosine kinase. Elife 2015, 4, e10592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanayama, A.; Seth, R.B.; Sun, L.; Ea, C.-K.; Hong, M.; Shaito, A.; Chiu, Y.-H.; Deng, L.; Chen, Z.J. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell. 2004, 15, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the Iκb kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Manning, E.; Pullen, S.S.; Souza, D.J.; Kehry, M.; Noelle, R.J. Cellular responses to murine CD40 in a mouse B cell line may be TRAF dependent or independent. Eur. J. Immunol. 2002, 32, 39–49. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M.; et al. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-γ that regulates neoangiogenesis. FASEB J. 2015, 29, 3638–3653. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S.-I. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 2003, 278, 46832–46839. [Google Scholar] [CrossRef] [Green Version]
- Sankala, H.M.; Hait, N.C.; Paugh, S.W.; Shida, D.; Lépine, S.; Elmore, L.W.; Dent, P.; Milstien, S.; Spiegel, S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007, 67, 10466–10474. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Sonoda, H.; Yu, H.; Kajimoto, T.; Goparaju, S.K.; Jahangeer, S.; Okada, T.; Nakamura, S.-I. Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J. Biol. Chem. 2007, 282, 27493–27502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; Maczis, M.A.; Maceyka, M.; Milstien, S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J. Lipid Res. 2019, 60, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the mucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Jamaladdin, S.; Kelly, R.D.W.; O’Regan, L.; Dovey, O.M.; Hodson, G.E.; Millard, C.J.; Portolano, N.; Fry, A.M.; Schwabe, J.W.R.; Cowley, S.M. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9840–9845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.D.W.; Chandru, A.; Watson, P.J.; Song, Y.; Blades, M.; Robertson, N.S.; Jamieson, A.G.; Schwabe, J.W.R.; Cowley, S.M. Histone deacetylase (HDAC) 1 and 2 complexes regulate both histone acetylation and crotonylation in vivo. Sci. Rep. 2018, 8, 14690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skoralakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Park, S.-M.; Kang, M.R.; Oh, S.-Y.; Lee, T.H.; Muller, M.T.; Chung, I.K. Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes Dev. 2005, 19, 776–781. [Google Scholar] [CrossRef] [Green Version]
- Murata, N.; Sato, K.; Kon, J.; Tomura, H.; Yanagita, M.; Kuwabara, A.; Ui, M.; Okajima, F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem. J. 2000, 352, 809–815. [Google Scholar] [CrossRef]
- Argraves, K.M.; Argraves, W.S. HDL serves as a S1P signaling platform mediating a multitude of cardiovascular effects. J. Lipid Res. 2007, 48, 2325–2333. [Google Scholar] [CrossRef] [Green Version]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurano, M.; Tsukamoto, K.; Ohkawa, R.; Hara, M.; Iino, J.; Kageyama, Y.; Ikeda, H.; Yatomi, Y. Liver involvement in sphingosine 1-phosphate dynamism revealed by adenoviral hepatic overexpression of apolipoprotein M. Atherosclerosis. 2013, 229, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Seo, J.; Allegood, J.; Bi, X.; Zhu, X.; Boudyguina, E.; Gebre, A.K.; Avni, D.; Shah, D.; Sorci-Thomas, M.G.; et al. Hepatic apolipoprotein M (ApoM) overexpression stimulates formation of larger ApoM/sphingosine 1-phosphate-enriched plasma high density lipoprotein. J. Biol. Chem. 2014, 289, 2801–2814. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T.; Estrada-Hernandez, T.; Paik, J.-H.; Wu, M.-T.; Venkataraman, K.; Brinkmann, V.; Claffey, K.; Hla, T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J. Biol. Chem. 2003, 278, 47281–47290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical profiling of G protein-coupled receptor expression. Cell 2008, 135, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Rosen, H.; Goetzl, E.J. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat. Rev. Immunol. 2005, 5, 560–570. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 2007, 18, 300–307. [Google Scholar] [CrossRef]
- Rosen, H.; Stevens, R.C.; Hanson, M.; Roberts, E.; Oldstone, M.B.A. Sphingosine-1-phosphate and its receptors: Structure, signaling, and influence. Annu. Rev. Biochem. 2013, 82, 637–662. [Google Scholar] [CrossRef]
- Badawy, S.M.M.; Okada, T.; Kajimoto, T.; Ijuin, T.; Nakamura, S.I. DHHC5-mediated palmitoylation of S1P receptor subtype 1 determines G-protein coupling. Sci. Rep. 2017, 7, 16552. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Tucker, A.E.; Tran, J.; Bergner, J.B.; Turner, E.M.; Proia, R.L. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J. Clin. Invest. 2014, 124, 2076–2086. [Google Scholar] [CrossRef] [Green Version]
- Kono, M.; Conlon, E.G.; Lux, S.Y.; Yanagida, K.; Hla, T.; Proia, R.L. Bioluminescence imaging of G protein-coupled receptor activation in living mice. Nat. Commun. 2017, 8, 1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnon, T.I.; Xu, Y.; Lo, C.; Pham, T.; An, J.; Coughlin, S.; Dorn, G.W.; Cyster, J.G. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science 2011, 333, 1898–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangada, S.; Khanna, K.M.; Blaho, V.A.; Oo, M.L.; Im, D.-S.; Guo, C.; Lefrancois, L.; Hla, T. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J. Exp. Med. 2010, 207, 1475–1483. [Google Scholar] [CrossRef]
- Willinger, T.; Ferguson, S.M.; Pereira, J.P.; De Camilli, P.; Flavell, R.A. Dynamin 2-dependent endocytosis is required for sustained S1PR1 signaling. J. Exp. Med. 2014, 211, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Nomachi, A.; Yoshinaga, M.; Liu, J.; Kanchanawong, P.; Tohyama, K.; Thumkeo, D.; Watanabe, T.; Narumiya, S.; Hirata, T. Moesin controls clathrin-mediated S1PR1 internalization in T cells. PLoS ONe 2013, 8, e82590. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.H.; Thangada, S.; Lee, M.J.; Van Brooklyn, J.R.; Spiegel, S.; Hla, T. Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG- 1. Mol. Biol. Cell. 1999, 10, 1179–1190. [Google Scholar] [CrossRef] [Green Version]
- Oo, M.L.; Chang, S.H.; Thangada, S.; Wu, M.-T.; Rezaul, K.; Blaho, V.; Hwang, S.-I.; Han, D.K.; Hla, T. Engagement of S1P1-degradative mechanisms leads to vascular leak in mice. J. Clin. Invest. 2011, 121, 2290–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, H.; Kolter, T.; Sandhoff, K. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 2009, 1793, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzio, J.P.; Gray, S.R.; Bright, N.A. Endosome-lysosome fusion. Biochem. Soc. Trans. 2010, 38, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Invest. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.M.; Takahashi, Y.; Fox, T.E.; Yun, J.K.; Kester, M.; Wang, H.-G. Sphingosine kinase 1 cooperates with autophagy to maintain endocytic membrane trafficking. Cell Rep. 2016, 17, 1532–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Hla, T.; Lynch, K.R.; Spiegel, S.; Moolenaar, W.H. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2010, 62, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.; Han, J.E.; Jeon, S.; Ryu, J.H.; Choi, J.W.; Chun, J. Exogenous S1P exposure potentiates ischemic stroke damage that is reduced possibly by inhibiting S1P receptor signaling. Mediators Inflamm. 2015, 2015, 492659. [Google Scholar] [CrossRef]
- Siehler, S.; Manning, D.R. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochim. Biophys. Acta 2002, 1582, 94–99. [Google Scholar] [CrossRef]
- Gonda, K.; Okamoto, H.; Takuwa, N.; Yatomi, Y.; Okazaki, H.; Sakurai, T.; Kimura, S.; Sillard, R.; Harii, K.; Takuwa, Y. The novel sphingosine 1-phosphate receptor AGR16 is coupled via pertussis toxin-sensitive and -insensitive G-proteins to multiple signalling pathways. Biochem. J. 1999, 337, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Windh, R.T.; Lee, M.J.; Hla, T.; An, S.; Barr, A.J.; Manning, D.R. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G12 families of heterotrimeric G proteins. J. Biol. Chem. 1999, 274, 27351–27358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, H.; Takuwa, N.; Yokomizo, T.; Sugimoto, N.; Sakurada, S.; Shigematsu, H.; Takuwa, Y. Inhibitory regulation of Rac activation, membrane ruffling, and cell migration by the G protein-coupled sphingosine-1-phosphate receptor EDG5 but not EDG1 or EDG3. Mol. Cell Biol. 2000, 20, 9247–9261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ancellin, N.; Hla, T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J. Biol. Chem. 1999, 274, 18997–19002. [Google Scholar] [CrossRef] [Green Version]
- Graeler, M.; Goetzl, E.J. Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. FASEB J. 2002, 16, 1874–1878. [Google Scholar] [CrossRef] [Green Version]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Ghosh, P.; Rangamani, P.; Kufareva, I. The GAPs, GEFs, GDIs and…now, GEMs: New kids on the heterotrimeric G protein signaling block. Cell Cycle. 2017, 16, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Feistritzer, C.; Riewald, M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 2005, 105, 3178–3184. [Google Scholar] [CrossRef]
- Singleton, P.A.; Dudek, S.M.; Ma, S.-F.; Garcia, J.G.N. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation: Novel role for hyaluronan and CD44 receptor family. J. Biol. Chem. 2006, 281, 34381–34393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiow, L.R.; Rosen, D.B.; Brdičková, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Kono, M.; Cartier, A.; Engelbrecht, E.; Kano, K.; Kawakami, K.; Xiong, Y.; Piao, W.; Galvani, S.; Yanagida, K.; et al. Lysolipid receptor cross-talk regulates lymphatic endothelial junctions in lymph nodes. J. Exp. Med. 2019, 216, 1582–1598. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Gray, E.E.; Zhang, Y.; Ramírez-Valle, F.; Cyster, J.G. Sphingosine-1-phosphate receptor 2 restrains egress of γδ T cells from the skin. J. Exp. Med. 2019, 216, 1487–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, A.; Tews, B.; Arzt, M.E.; Weinmann, O.; Obermair, F.J.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The sphingolipid receptor S1PR2 is a receptor for Nogo-A repressing synaptic plasticity. PLoS Biol. 2014, 12, e1001763. [Google Scholar] [CrossRef]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, D.; Zhang, Y.; Wang, P.; Zheng, C.; Zhang, S.; Yu, B.; Zhang, L.; Zhao, G.; Ma, B.; et al. Novel adipokine, FAM19A5, inhibits neointima formation after injury through sphingosine-1-phosphate receptor 2. Circulation 2018, 138, 48–63. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Angot, E.; Steiner, J.A.; Hansen, C.; Li, J.-Y.; Brundin, P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010, 9, 1128–1138. [Google Scholar] [CrossRef]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Shubhra Chakrabarti, S.; Bir, A.; Poddar, J.; Sinha, M.; Ganguly, A.; Chakrabarti, S. Ceramide and sphingosine-1-phosphate in cell death pathways: Relevance to the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1232–1248. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Van Echten-Deckert, G.; Walter, J. Sphingolipids: Critical players in Alzheimer’s disease. Prog. Lipid Res. 2012, 51, 378–393. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Bevan, D.R. Lipid composition influences the release of Alzheimer’s amyloid β-peptide from membranes. Protein Sci. 2011, 20, 1530–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid rafts and neurodegeneration: Structural and functional roles in physiologic aging and neurodegenerative diseases. J. Lipid Res. 2020, 61, 636–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crivelli, S.M.; Giovagnoni, C.; Visseren, L.; Scheithauer, A.-L.; de Wit, N.; den Hoedt, S.; Losen, M.; Mulder, M.T.; Walter, J.; de Vries, H.E. et al.; et al. Sphingolipids in Alzheimer’s disease, how can we target them? Adv. Drug Deliv. Rev 2020, S0169-409X(20)30002-8. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Cairns, N.J.; Da, X.; Chen, K.; Carter, D.; Fleisher, A.; Householder, E.; Ayutyanont, N.; Roontiva, A.; Bauer, R.J.; et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol. Commun. 2013, 1, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider. JA. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Becker, K.A.; Fahsel, B.; Kemper, H.; Mayeres, J.; Li, C.; Wilker, B.; Keitsch, S.; Soddemann, M.; Sehl, C.; Kohnen, M.; et al. Staphylococcus aureus alpha-toxin disrupts endothelial-cell tight junctions via acid sphingomyelinase and ceramide. Infect. Immun. 2017, 86, e00606–17. [Google Scholar] [CrossRef] [Green Version]
- van Doorn, R.; Nijland, P.G.; Dekker, N.; Witte, M.E.; Lopes-Pinheiro, M.A.; van het Hof, B.; Kooij, G.; Reijerkerk, A.; Dijkstra, C.; van van der Valk, P.; et al. Fingolimod attenuates ceramide-induced blood-brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012, 124, 397–410. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.-X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with alzheimer’s and other neurodegenerative diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couttas, T.A.; Kain, N.; Daniels, B.; Lim, X.Y.; Shepherd, C.; Kril, J.; Pickford, R.; Li, H.; Garner, B.; Don, A.S. Loss of the neuroprotective factor sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2014, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Katsel, P.; Li, C.; Haroutunian, V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 12. [Google Scholar] [CrossRef]
- Dominguez, G.; Maddelein, M.-L.; Pucelle, M.; Nicaise, Y.; Maurage, C.-A.; Duyckaerts, C.; Cuvillier, O.; Delisle, M.-B. Neuronal sphingosine kinase 2 subcellular localization is altered in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2018, 6, 25. [Google Scholar] [CrossRef]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Stefanis, L. α-synuclein degradation by autophagic pathways: A potential key to Parkinson’s disease pathogenesis. Autophagy 2008, 4, 917–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef]
- Manzoni, C.; Lewis, P.A. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 2013, 27, 3424–3429. [Google Scholar] [CrossRef] [Green Version]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Samii, A.; Schneer, S.H.; Roberts, J.W.; Griffith, A.; Leis, B.C.; Schellenberg, G.D.; Sidransky, E.; Bird, T.D.; Leverenz, J.B.; et al. Glucocerebrosidase gene mutations: A risk factor for Lewy body disorders. Arch. Neurol. 2008, 65, 379–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bras, J.; Singleton, A.; Cookson, M.R.; Hardy, J. Emerging pathways in genetic Parkinson’s disease: Potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008, 275, 5767–5773. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease and parkinsonism. Mol. Genet. Metab. 2005, 84, 302–304. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated parkinson’s disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [Green Version]
- Everett, C.M.; Wood, N.W. Trinucleotide repeats and neurodegenerative disease. Brain 2004, 127, 2385–2405. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef] [Green Version]
- Di Pardo, A.; Amico, E.; Basit, A.; Armirotti, A.; Joshi, P.; Neely, M.D.; Vuono, R.; Castaldo, S.; Digilio, A.F.; Scalabrì, F.; et al. Defective sphingosine-1-phosphate metabolism is a druggable target in Huntington’s disease. Sci. Rep. 2017, 7, 5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirhaji, L.; Milani, P.; Dalin, S.; Wassie, B.T.; Dunn, D.E.; Fenster, R.J.; Avila-Pacheco, J.; Greengard, P.; Clish, C.B.; Heiman, M.; et al. Identifying therapeutic targets by combining transcriptional data with ordinal clinical measurements. Nat. Commun. 2017, 8, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pardo, A.; Maglione, V. The S1P axis: New exciting route for treating Huntington’s disease. Trends Pharmacol. Sci. 2018, 39, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.-P. Sphingolipid metabolism is dysregulated at transcriptomic and metabolic levels in the spinal cord of an animal model of amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.-J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef]
- Potenza, R.L.; De Simone, R.; Armida, M.; Mazziotti, V.; Pèzzola, A.; Popoli, P.; Minghetti, L. Fingolimod: A disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics 2016, 13, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Grant, J.L.; Ghosn, E.E.B.; Axtell, R.C.; Herges, K.; Kuipers, H.F.; Woodling, N.S.; Andreasson, K.; Herzenberg, L.A.; Herzenberg, L.A.; Steinman, L. Reversal of paralysis and reduced inflammation from peripheral administration of β-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci. Transl. Med. 2012, 4, 145ra105. [Google Scholar] [CrossRef] [Green Version]
- Gijbels, K.; Engelborghs, S.; De Deyn, P.P. Experimental autoimmune encephalomyelitis: An animal model for multiple sclerosis. Neurosci. Res. Commun. 2000, 26, 193–206. [Google Scholar] [CrossRef]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M.; Funeshima, N.; Kitazawa, Y.; Kimura, H.; Amemiya, H.; Suzuki, S.; Li, X.-K. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J. Pharmacol. Exp. Ther. 2003, 305, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.; Tham, C.-S.; Lin, F.-F.; Lariosa-Willingham, K.; Yu, N.; Hale, J.; Mandala, S.; Chun, J.; Rao, T.S. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J. Neuroimmunol. 2004, 153, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Sugahara, K.; Shimano, K.; Teshima, K.; Koyama, M.; Fukunari, A.; Chiba, K. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol. Immunol. 2005, 2, 439–448. [Google Scholar]
- Foster, C.A.; Mechtcheriakova, D.; Storch, M.K.; Balatoni, B.; Howard, L.M.; Bornancin, F.; Wlachos, A.; Sobanov, J.; Kinnunen, A.; Baumruker, T. FTY720 rescue therapy in the dark agouti rat model of experimental autoimmune encephalomyelitis: Expression of central nervous system genes and reversal of blood-brain-barrier damage. Brain Pathol. 2009, 19, 254–266. [Google Scholar] [CrossRef]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Eken, A.; Duhen, R.; Singh, A.K.; Fry, M.; Buckner, J.H.; Kita, M.; Bettelli, E.; Oukka, M. S1P1 deletion differentially affects TH17 and regulatory T cells. Sci. Rep. 2017, 7, 12905. [Google Scholar] [CrossRef] [Green Version]
- Smigiel, K.S.; Richards, E.; Srivastava, S.; Thomas, K.R.; Dudda, J.C.; Klonowski, K.D.; Campbell, D.J. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med. 2014, 211, 121–136. [Google Scholar] [CrossRef]
- Cruz-Orengo, L.; Daniels, B.P.; Dorsey, D.; Basak, S.A.; Grajales-Reyes, J.G.; McCandless, E.E.; Piccio, L.; Schmidt, R.E.; Cross, A.H.; Crosby, S.D.; et al. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J. Clin. Invest. 2014, 124, 2571–2584. [Google Scholar] [CrossRef] [Green Version]
- Lopes Pinheiro, M.A.; Kroon, J.; Hoogenboezem, M.; Geerts, D.; van Het Hof, B.; van der Pol, S.M.A.; van Buul, J.D.; de Vries, H.E. Acid sphingomyelinase–derived ceramide regulates ICAM-1 function during T cell transmigration across brain endothelial cells. J. Immunol. 2016, 196, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, F.; Schaffner, F.; Furlan-Freguia, C.; Pawlinski, R.; Bhattacharjee, G.; Chun, J.; Derian, C.K.; Andrade-Gordon, P.; Rosen, H.; Ruf, W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 2008, 452, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS One. 2011, 6, e23905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflammation 2017, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Devic, E. Congrès français de médecine (Premiere Session; Lyon, 1894; procès-verbaux, mémoires et discussions; publiés par M. le Dr, L. Bard); Asselin et Houzeau: Lyon, France, 1895. [Google Scholar]
- Gault, F. De la neuromyélite optique aiguë. Ph.D. Thesis, Faculté de Médecine Lyon Est, Lyon, France, 1894. [Google Scholar]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Weinshenker, B.G.; Wingerchuk, D.M.; Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A. NMO-IgG: A specific biomarker for neuromyelitis optica. Dis. Markers 2006, 22, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Franciotta, D.; Bergamaschi, R.; Wright, H.; Littleton, E.; Palace, J.; Hohlfeld, R.; Vincent, A. NMO-IgG in the diagnosis of neuromyelitis optica. Neurology 2007, 68, 1076–1077. [Google Scholar] [CrossRef]
- Min, J.H.; Kim, B.J.; Lee, K.H. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult. Scler. 2012, 18, 113–115. [Google Scholar] [CrossRef]
- Yoshii, F.; Moriya, Y.; Ohnuki, T.; Ryo, M.; Takahashi, W. Fingolimod-induced leukoencephalopathy in a patient with neuromyelitis optica spectrum disorder. Mult. Scler. Relat Disord. 2016, 7, 53–57. [Google Scholar] [CrossRef]
- Izaki, S.; Narukawa, S.; Kubota, A.; Mitsui, T.; Fukaura, H.; Nomura, K. A case of neuromyelitis optica spectrum disorder developing a fulminant course with multiple white-matter lesions following fingolimod treatment. Clin. Neurol. 2013, 53, 513–517. [Google Scholar] [CrossRef]
- Tanaka, M.; Oono, M.; Motoyama, R.; Tanaka, K. Longitudinally extensive spinal cord lesion after initiation, and multiple extensive brain lesions after cessation of fingolimod treatment in a patient with recurrent myelitis and anti-aquaporin 4 antibodies. Clin. Exp. Neuroimmunol. 2013, 4, 239–240. [Google Scholar] [CrossRef]
- Matsushita, T.; Tateishi, T.; Isobe, N.; Yonekawa, T.; Yamasaki, R.; Matsuse, D.; Murai, H.; Kira, J.-I. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One 2013, 8, e61835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.-C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.V.; Wu, Y.Y.; Liu, Q.; Wang, D.; Nguyen, S.; Loh, R.; Pang, J.; Friedman, K.; Orlofsky, A.; Augenlicht, L.; et al. STAT3 in epithelial cells regulates inflammation and tumor progression to malignant state in colon. Neoplasia 2013, 15, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Jackson, H.; Panopoulos, A.D.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF-induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood 2010, 115, 3354–3363. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, R.M.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.A.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef] [Green Version]
- Yopp, A.C.; Ochando, J.C.; Mao, M.; Ledgerwood, L.; Ding, Y.; Bromberg, J.S. Sphingosine 1-phosphate receptors regulate chemokine-driven transendothelial migration of lymph node but not splenic T cells. J. Immunol. 2005, 175, 2913–2924. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Yester, J.W.; Surace, M.J.; Oyeniran, C.; Price, M.M.; Huang, W.-C.; Hait, N.C.; Allegood, J.C.; Yamada, A.; Kong, X.; et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat. Immunol. 2014, 15, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Uzawa, A.; Mori, M.; Arai, K.; Sato, Y.; Hayakawa, S.; Masuda, S.; Taniguchi, J.; Kuwabara, S. Cytokine and chemokine profiles in neuromyelitis optica: Significance of interleukin-6. Mult. Scler. 2010, 16, 1443–1452. [Google Scholar] [CrossRef]
- Muscal, E.; Brey, R.L. Neurologic manifestations of systemic lupus erythematosus in children and adults. Neurol. Clin. 2010, 28, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirshner, H.S. Hashimoto’s encephalopathy: A brief review. Curr. Neurol. Neurosci. Rep. 2014, 14, 476. [Google Scholar] [CrossRef]
- Sanna, G.; Piga, M.; Terryberry, J.W.; Peltz, M.T.; Giagheddu, S.; Satta, L.; Ahmed, A.; Cauli, A.; Montaldo, C.; Passiu, G.; et al. Central nervous system involvement in systemic lupus erythematosus: Cerebral imaging and serological profile in patients with and without overt neuropsychiatric manifestations. Lupus 2000, 9, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Snider, A.J. Sphingosine kinase and sphingosine-1-phosphate: Regulators in autoimmune and inflammatory disease. Int. J. Clin. Rheumtol. 2013, 8, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, H.; Hirata, D.; Kamimura, T.; Sato, H.; Iwamoto, M.; Yoshio, T.; Masuyama, J.; Fujimura, A.; Kobayashi, E.; Kano, S.; et al. Effects of FTY720 in MRL-lpr/lpr mice: Therapeutic potential in systemic lupus erythematosus. J. Rheumatol. 2002, 29, 707–716. [Google Scholar] [PubMed]
- Alperovich, G.; Rama, I.; Lloberas, N.; Franquesa, M.; Poveda, R.; Gomà, M.; Herrero-Fresneda, I.; Cruzado, J.M.; Bolaños, N.; Carrera, M.; et al. New immunosuppresor strategies in the treatment of murine lupus nephritis. Lupus 2007, 16, 18–24. [Google Scholar] [CrossRef]
- Ando, S.; Amano, H.; Amano, E.; Minowa, K.; Watanabe, T.; Nakano, S.; Nakiri, Y.; Morimoto, S.; Tokano, Y.; Lin, Q.; et al. FTY720 exerts a survival advantage through the prevention of end-stage glomerular inflammation in lupus-prone BXSB mice. Biochem. Biophys. Res. Commun. 2010, 394, 804–810. [Google Scholar] [CrossRef]
- Wenderfer, S.E.; Stepkowski, S.M.; Braun, M.C. Increased survival and reduced renal injury in MRL/lpr mice treated with a novel sphingosine-1-phosphate receptor agonist. Kidney Int. 2008, 74, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; He, X.; Xia, X.; Guo, J.; Liu, A.; Liu, X.; Wang, X.; Li, C.; Peng, S.; Zhao, W.; et al. Sphk1/S1P/S1PR1 Signaling is involved in the development of autoimmune thyroiditis in patients and NOD.H-2h4 mice. Thyroid 2019, 29, 700–713. [Google Scholar] [CrossRef]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Lo, E.H. T time in the brain. Nat. Med. 2009, 15, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Hurn, P.D.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Kaler, L.J.; Vandenbark, A.A.; Offner, H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J. Cereb. Blood Flow Metab. 2007, 27, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedor, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef]
- Adachi, K.; Kohara, T.; Nakao, N.; Arita, M.; Chiba, K.; Mishina, T.; Sazaki, S.; Fujita, T. Design, synthesis, and structure-activity relationships of 2-substituted-2-amino-1,3-propanediols: Discovery of a novel immunosuppressant, FTY720. Bioorganic Med. Chem. Lett. 1995, 5, 853–856. [Google Scholar] [CrossRef]
- Brinkmann, V.; Cyster, J.G.; Hla, T. FTY720: Sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am. J. Transplant. 2004, 4, 1019–1025. [Google Scholar] [CrossRef]
- Rolland, W.B., II.; Manaenko, A.; Lekic, T.; Hasegawa, Y.; Ostrowski, R.; Tang, J.; Zhang, J.H. FTY720 is neuroprotective and improves functional outcomes after intracerebral hemorrhage in mice. Acta Neurochir. Suppl. 2011, 111, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, Y.; Suzuki, H.; Sozen, T.; Rolland, W.; Zhang, J.H. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 2010, 41, 368–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, B.; Pfeilschifter, W.; Mazaheri-Omrani, N.; Strobel, M.A.; Kahles, T.; Neumann-Haefelin, T.; Rami, A.; Huwiler, A.; Pfeilschifter, J. The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem. Biophys. Res. Commun. 2009, 389, 251–256. [Google Scholar] [CrossRef]
- Kraft, P.; Göb, E.; Schuhmann, M.K.; Göbel, K.; Deppermann, C.; Thielmann, I.; Herrmann, A.M.; Lorenz, K.; Brede, M.; Stoll, G.; et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke 2013, 44, 3202–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazari, M.; Keshavarz, S.; Rafati, A.; Namavar, M.R.; Haghani, M. Fingolimod (FTY720) improves hippocampal synaptic plasticity and memory deficit in rats following focal cerebral ischemia. Brain Res. Bull. 2016, 124, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yemisci, M.; Kim, H.-H.; Yung, L.M.; Shin, H.K.; Hwang, S.-K.; Guo, S.; Qin, T.; Alsharif, N.; Brinkmann, V.; et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011, 69, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeilschifter, W.; Czech-Zechmeister, B.; Sujak, M.; Mirceska, A.; Koch, A.; Rami, A.; Steinmetz, H.; Foerch, C.; Huwiler, A.; Pfeilschifter, J. Activation of sphingosine kinase 2 is an endogenous protective mechanism in cerebral ischemia. Biochem. Biophys. Res. Commun. 2011, 413, 212–217. [Google Scholar] [CrossRef]
- Rolland, W.B.; Lekic, T.; Krafft, P.R.; Hasegawa, Y.; Altay, O.; Hartman, R.; Ostrowski, R.; Manaenko, A.; Tang, J.; Zhang, J.H. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp. Neurol. 2013, 241, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Zhang, N.; Ren, L.; Yan, Y.; Sun, N.; Li, Y.-J.; Han, W.; Xue, R.; Liu, Q.; Hao, J.; et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc. Natl. Acad. Sci. USA 2014, 111, 18315–18320. [Google Scholar] [CrossRef] [Green Version]
- Schaphorst, K.L.; Chiang, E.; Jacobs, K.N.; Zaiman, A.; Natarajan, V.; Wigley, F.; Garcia, J.G.N. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L258–L267. [Google Scholar] [CrossRef] [Green Version]
- Liesz, A.; Zhou, W.; Mracskó, É.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegemann, S.; Cerwenka, A.; et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 2011, 134, 704–720. [Google Scholar] [CrossRef] [Green Version]
- Cai, A.; Schlunk, F.; Bohmann, F.; Kahefiolasl, S.; Brunkhorst, R.; Foerch, C.; Pfeilschifter, W. Coadministration of FTY720 and rt-PA in an experimental model of large hemispheric stroke-No influence on functional outcome and blood-brain barrier disruption. Exp. Transl. Stroke Med. 2013, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T. Sphingosine-1-phosphate signaling in endothelial disorders. Curr. Atheroscler. Rep. 2016, 18, 31. [Google Scholar] [CrossRef]
- Brait, V.H.; Tarrasón, G.; Gavaldà, A.; Godessart, N.; Planas, A.M. Selective sphingosine 1-phosphate receptor 1 agonist is protective against ischemia/reperfusion in mice. Stroke 2016, 47, 3053–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondeau, N.; Lai, Y.; Tyndall, S.; Popolo, M.; Topalkara, K.; Pru, J.K.; Zhang, L.; Kim, H.; Liao, J.K.; Ding, K.; et al. Distribution of sphingosine kinase activity and mRNA in rodent brain. J. Neurochem. 2007, 103, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billich, A.; Bornancin, F.; Dévay, P.; Mechtcheriakova, D.; Urtz, N.; Baumruker, T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J. Biol. Chem. 2003, 278, 47408–47415. [Google Scholar] [CrossRef] [Green Version]
- Wacker, B.K.; Perfater, J.L.; Gidday, J.M. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J. Neurochem. 2012, 123, 954–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemann, B.; Kinzel, B.; Müller, M.; Reuschel, R.; Mechtcheriakova, D.; Urtz, N.; Bornancin, F.; Baumruker, T.; Billich, A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 2006, 107, 1454–1458. [Google Scholar] [CrossRef]
- Wacker, B.K.; Park, T.S.; Gidday, J.M. Hypoxic preconditioning-induced cerebral ischemic tolerance: Role of microvascular sphingosine kinase 2. Stroke 2009, 40, 3342–3348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; An, J.; Jawadi, H.; Siow, D.L.; Lee, J.-F.; Zhao, J.; Gartung, A.; Maddipati, K.R.; Honn, K.V.; Wattenberg, B.W.; et al. Sphingosine-1-phosphate receptor-2 mediated NFκB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 2013, 106, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.S.; Yang, L.; Zhang, G.; Zhao, H.; Selim, M.; McCullough, L.D.; Kluk, M.J.; Sanchez, T. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat. Commun. 2015, 6, 7893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.; Zhang, D.; Dai, D.; Zhang, W.; Zhang, L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. 2016, 65, 551–562. [Google Scholar] [CrossRef]
- Zheng, S.; Wei, S.; Wang, X.; Xu, Y.; Xiao, Y.; Liu, H.; Jia, J.; Cheng, J. Sphingosine kinase 1 mediates neuroinflammation following cerebral ischemia. Exp. Neurol. 2015, 272, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.; Qin, T.; Castillo, J.; Seo, J.H.; Arai, K.; Lo, E.H.; Waeber, C. Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke 2013, 44, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Fu, Y.; Tian, D.; Sun, N.; Han, W.; Chang, G.; Dong, Y.; Xu, X.; Liu, Q.; Huang, D.; et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: A pilot trial. Circulation 2015, 132, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhou, Y.; Zhang, R.; Zhang, M.; Campbell, B.; Lin, L.; Shi, F.-D.; Lou, M. Rationale and design of combination of an immune modulator fingolimod with alteplase bridging with mechanical thrombectomy in acute ischemic stroke (FAMTAIS) trial. Int. J. Stroke 2017, 12, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Jin, H.-J.; Zhu, Y.-Y.; Fang, Z.; Mao, L.; He, Q.; Xia, J.-P.; Li, M.; Li, Y.; Chen, X.; et al. MicroRNA-149-5p regulates blood-brain barrier permeability after transient middle cerebral artery occlusion in rats by targeting S1PR2 of pericytes. FASEB J. 2018, 32, 3133–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swendeman, S.L.; Xiong, Y.; Cantalupo, A.; Yuan, A.; Burg, N.; Hisano, Y.; Cartier, A.; Liu, C.H.; Engelbrecht, E.; Blaho, V.; et al. An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci. Signal. 2017, 10, eaal2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testai, F.D.; Kilkus, J.P.; Berdyshev, E.; Gorshkova, I.; Natarajan, V.; Dawson, G. Multiple sphingolipid abnormalities following cerebral microendothelial hypoxia. J. Neurochem. 2014, 131, 530–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.-G.; Choi, J.W. Identification of sphingosine 1-phosphate receptor subtype 1 (S1P1) as a pathogenic factor in transient focal cerebral ischemia. Mol. Neurobiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]
- Gaire, B.P.; Song, M.-R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflammation 2018, 15, 284. [Google Scholar] [CrossRef]
- Gaire, B.P.; Bae, Y.J.; Choi, J.W. S1P1 regulates M1/M2 polarization toward brain injury after transient focal cerebral ischemia. Biomol. Ther. 2019, 27, 522–529. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Bassi, R.; Tettamanti, G.; Viani, P.; Riboni, L. Extracellular release of newly synthesized sphingosine-1-phosphate by cerebellar granule cells and astrocytes. J. Neurochem. 2005, 92, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Craveiro, L.M.; Shin, Y.; Cortes-Cros, M.; Bassilana, F.; Osinde, M.; Wishart, W.L.; Guerini, D.; Thallmair, M.; Schwab, M.E.; et al. Phosphorylated FTY720 promotes astrocyte migration through sphingosine-1-phosphate receptors. J. Neurochem. 2007, 102, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.R.; Chun, J. Effects of LPA and S1P on the nervous system and implications for their involvement in disease. Curr. Drug Targets 2007, 8, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef]
- Yagi, K.; Lidington, D.; Wan, H.; Fares, J.C.; Meissner, A.; Sumiyoshi, M.; Ai, J.; Foltz, W.D.; Nedospasov, S.A.; Offermanns, S.; et al. Therapeutically targeting tumor necrosis factor-α/sphingosine-1-phosphate signaling corrects myogenic reactivity in subarachnoid hemorrhage. Stroke 2015, 46, 2260–2270. [Google Scholar] [CrossRef] [Green Version]
- Olsson, T.; Zhi, W.W.; Hojeberg, B.; Kostulas, V.; Jiang, Y.P.; Anderson, G.; Ekre, H.P.; Link, H. Autoreactive T lymphocytes in multiple sclerosis determined by antigen-induced secretion of interferon-γ. J. Clin. Invest. 1990, 86, 981–985. [Google Scholar] [CrossRef]
- Comi, G. Position and practical use of fingolimod in Europe. Clin. Exp. Neuroimmunol. 2014, 5, 19–33. [Google Scholar] [CrossRef]
- Camm, J.; Hla, T.; Bakshi, R.; Brinkmann, V. Cardiac and vascular effects of fingolimod: Mechanistic basis and clinical implications. Am. Heart J. 2014, 168, 632–644. [Google Scholar] [CrossRef]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Khatri, B.O.; Montalba, Y.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Arvin, A.M.; Wolinsky, J.S.; Kappos, L.; Morris, M.I.; Reder, A.T.; Tornatore, C.; Gershon, A.; Gershon, M.; Levin, M.J.; Bezuidenhoudt, M.; et al. Varicella-zoster virus infections in patients treated with fingolimod: Risk assessment and consensus recommendations for management. JAMA Neurol. 2015, 72, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerch, C.; Friedauer, L.; Bauer, B.; Wolf, T.; Adam, E.H. Severe COVID-19 infection in a patient with multiple sclerosis treated with fingolimod. Mult. Scler. Relat. Disord. 2020, 42, 102180. [Google Scholar] [CrossRef]
- Brinkmann, V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Merrill, A.H., Jr.; Dials, H.; Barbi, A.; Schwartz, G.K. A phase I clinical study of safingol followed by cisplatin: Promising activity in refractory adrenocortical cancer with novel pharmacology. J. Clin. Oncol. 2006, 24, 13044–13044. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Haimovitz-Friedman, A.; Dhupar, S.K.; Ehleiter, D.; Maslak, P.; Lai, L.; Loganzo, F., Jr.; Kelsen, D.P.; Fuks, Z.; Albino, A.P. Potentiation of apoptosis by treatment with the protein kinase C-specific inhibitor safingol in mitomycin C- treated gastric cancer cells. J. Natl. Cancer Inst. 1995, 87, 1394–1399. [Google Scholar] [CrossRef]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H., Jr.; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. 2011, 17, 2484–2492. [Google Scholar] [CrossRef] [Green Version]
- Ling, L.-U.; Tan, K.-B.; Lin, H.; Chiu, G.N.C. The role of reactive oxygen species and autophagy in safingol-induced cell death. Cell Death Dis. 2011, 2, e129. [Google Scholar] [CrossRef] [Green Version]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Summers, S.A. Ceramides - Lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [Green Version]
- Xun, C.; Chen, M.-B.; Qi, L.; Tie-Ning, Z.; Peng, X.; Ning, L.; Zhi-Xiao, C.; Li-Wei, W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Jin, L.; Yang, B.; Wang, L.; Xia, Z.; Zhang, Q.; Xu, J. The sphingosine kinase 2 inhibitor ABC294640 inhibits cervical carcinoma cell growth. Oncotarget 2018, 9, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Smith, C.D.; Foroozesh, M.; Miele, L.; Qin, Z. The sphingosine kinase 2 inhibitor ABC294640 displays anti-non-small cell lung cancer activities in vitro and in vivo. Int. J. Cancer 2018, 142, 2153–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [Green Version]
- Mahajan-Thakur, S.; Bien-Möller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme—A systematic review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef] [Green Version]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Caballero, S.; Swaney, J.; Moreno, K.; Afzal, A.; Kielczewski, J.; Stoller, G.; Cavalli, A.; Garland, W.; Hansen, G.; Sabbadini, R.; et al. Anti-sphingosine-1-phosphate monoclonal antibodies inhibit angiogenesis and sub-retinal fibrosis in a murine model of laser-induced choroidal neovascularization. Exp. Eye Res. 2009, 88, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Sabbadini, R.A. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br. J. Pharmacol. 2011, 162, 1225–1238. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef]
- Fleischmann, R. Novel small-molecular therapeutics for rheumatoid arthritis. Curr. Opin. Rheumatol. 2012, 24, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Pyszko, J.A.; Strosznajder, J.B. The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol. 2014, 52, 260–269. [Google Scholar] [CrossRef]
- Samuvel, D.J.; Saxena, N.; Dhindsa, J.S.; Singh, A.K.; Gill, G.S.; Grobelny, D.W.; Singh, I. AKP-11 - A novel S1P1 agonist with favorable safety profile attenuates experimental autoimmune encephalomyelitis in rat model of multiple sclerosis. PLoS ONE 2015, 10, e0141781. [Google Scholar] [CrossRef] [PubMed]
- Dhar, T.G.M.; Xiao, H.-Y.; Xie, J.; Lehman-McKeeman, L.D.; Wu, D.-R.; Dabros, M.; Yang, X.; Taylor, T.L.; Zhou, X.D.; Heimrich, E.M.; et al. Identification and preclinical pharmacology of BMS-986104: A differentiated S1P1 receptor modulator in clinical trials. ACS Med. Chem. Lett. 2016, 7, 283–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piali, L.; Birker-Robaczewska, M.; Lescop, C.; Froidevaux, S.; Schmitz, N.; Morrison, K.; Kohl, C.; Rey, M.; Studer, R.; Vezzali, E.; et al. Cenerimod, a novel selective S1P1 receptor modulator with unique signaling properties. Pharmacol. Res. Perspect. 2017, 5, e00370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, V.; Batalov, A.; Smakotina, S.; Juif, P.E.; Cornelisse, P. First use of cenerimod, a selective S1P 1 receptor modulator, for the treatment of SLE: A double-blind, randomised, placebo-controlled, proof-of-concept study. Lupus Sci. Med. 2019, 6, e000354. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Gray, F.; Henderson, A.; Hicks, K.; Yang, J.; Thompson, P.; Oliver, J. Safety, pharmacokinetics, pharmacodynamics, and bioavailability of GSK2018682, a sphingosine-1-phosphate receptor modulator, in healthy volunteers. Clin. Pharmacol. Drug Dev. 2014, 3, 170–178. [Google Scholar] [CrossRef]
- Gruessner, R.W.; Sutherland, D.E.; Troppmann, C.; Benedetti, E.; Hakim, N.; Dunn, D.L.; Gruessner, A.C. The surgical risk of pancreas transplantation in the cyclosporine era: An overview. J. Am. Coll. Surg. 1997, 185, 128–144. [Google Scholar] [CrossRef]
- Khattar, M.; Deng, R.; Kahan, B.D.; Schroder, P.M.; Phan, T.; Rutzky, L.P.; Stepkowski, S.M. Novel sphingosine-1-phosphate receptor modulator KRP203 combined with locally delivered regulatory T cells induces permanent acceptance of pancreatic islet allografts. Transplantation 2013, 95, 919–927. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, D.; Freedman, M.S.; Prinz, J. Ponesimod, a selective S1P1 receptor modulator: A potential treatment for multiple sclerosis and other immune-mediated diseases. Ther. Adv. Chronic Dis. 2016, 7, 18–33. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.G.; Herrero San Juan, M.; Trautmann, S.; Berninger, L.; Schwiebs, A.; Ottenlinger, F.M.; Thomas, D.; Zaucke, F.; Pfeilschifter, J.M.; Radeke, H.H. Sphingosine-1-phosphate receptor 5 modulates early-stage processes during fibrogenesis in a mouse model of systemic sclerosis: A pilot study. Front. Immunol. 2017, 8, 1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurata, H.; Kusumi, K.; Otsuki, K.; Suzuki, R.; Kurono, M.; Komiya, T.; Hagiya, H.; Mizuno, H.; Shioya, H.; Ono, T.; et al. Discovery of a 1-Methyl-3,4-dihydronaphthalene-based sphingosine-1-phosphate (S1P) receptor agonist ceralifimod (ONO-4641). A S1P1 and S1P5 selective agonist for the treatment of autoimmune diseases. J. Med. Chem. 2017, 60, 9508–9530. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.R.T.; Steinberg, M.W.; Clemons, B.; Stokes, M.E.; Opiteck, G.J.; Peach, R.; Scott, F.L. Ozanimod (RPC1063), a selective S1PR1 and S1PR5 modulator, reduces chronic inflammation and alleviates kidney pathology in murine systemic lupus erythematosus. PLoS ONE 2018, 13, e0193236. [Google Scholar] [CrossRef]
- Wang, W.; Graeler, M.H.; Goetzl, E.J. Type 4 sphingosine 1-phosphate G protein-coupled receptor (S1P4 ) transduces S1P effects on T cell proliferation and cytokine secretion without signaling migration. FASEB J. 2005, 19, 1731–1733. [Google Scholar] [CrossRef]
- Wang, W.; Huang, M.-C.; Goetzl, E.J. Type 1 sphingosine 1-phosphate G protein-coupled receptor (S1P1) mediation of enhanced IL-4 generation by CD4 T cells from S1P1 transgenic mice. J. Immunol. 2007, 178, 4885–4890. [Google Scholar] [CrossRef] [Green Version]
- Gräler, M.H.; Grosse, R.; Kusch, A.; Kremmer, E.; Gudermann, T.; Lipp, M. The sphingosine 1-phosphate receptor S1P4 regulates cell shape and motility via coupling to Gi and G12/13. J. Cell Biochem. 2003, 89, 507–519. [Google Scholar] [CrossRef]
- Schulze, T.; Golfier, S.; Tabeling, C.; Räbel, K.; Gräler, M.H.; Witzenrath, M.; Lipp, M. Sphingosine-1-phospate receptor 4 (S1P4) deficiency profoundly affects dendritic cell function and TH 17-cell differentiation in a murine model. FASEB J. 2011, 25, 4024–4036. [Google Scholar] [CrossRef]
- Sugahara, K.; Maeda, Y.; Shimano, K.; Mogami, A.; Kataoka, H.; Ogawa, K.; Hikida, K.; Kumagai, H.; Asayama, M.; Yamamoto, T.; et al. Amiselimod, a novel sphingosine 1-phosphate receptor-1 modulator, has potent therapeutic efficacy for autoimmune diseases, with low bradycardia risk. Br. J. Pharmacol. 2017, 174, 15–27. [Google Scholar] [CrossRef]
- Kappos, L.; Arnold, D.L.; Bar-Or, A.; Camm, A.J.; Derfuss, T.; Sprenger, T.; Davies, M.; Piotrowska, A.; Ni, P.; Harada, T. Two-year results from a phase 2 extension study of oral amiselimod in relapsing multiple sclerosis. Mult. Scler. 2018, 24, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Shimano, K.; Maeda, Y.; Kataoka, H.; Murase, M.; Mochizuki, S.; Utsumi, H.; Oshita, K.; Sugahara, K. Amiselimod (MT-1303), a novel sphingosine 1-phosphate receptor-1 functional antagonist, inhibits progress of chronic colitis induced by transfer of CD4+CD45RBhigh T cells. PLoS ONE 2019, 14, e0226154. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Zhang, J.; Chiorean, M.; Vermeire, S.; Lee, S.D.; Kühlbacher, T.; Yacyshyn, B.; Cabell, C.H.; Naik, S.U.; et al. Efficacy and safety of etrasimod in a phase 2 randomized trial of patients with ulcerative colitis. Gastroenterology 2020, 158, 550–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]




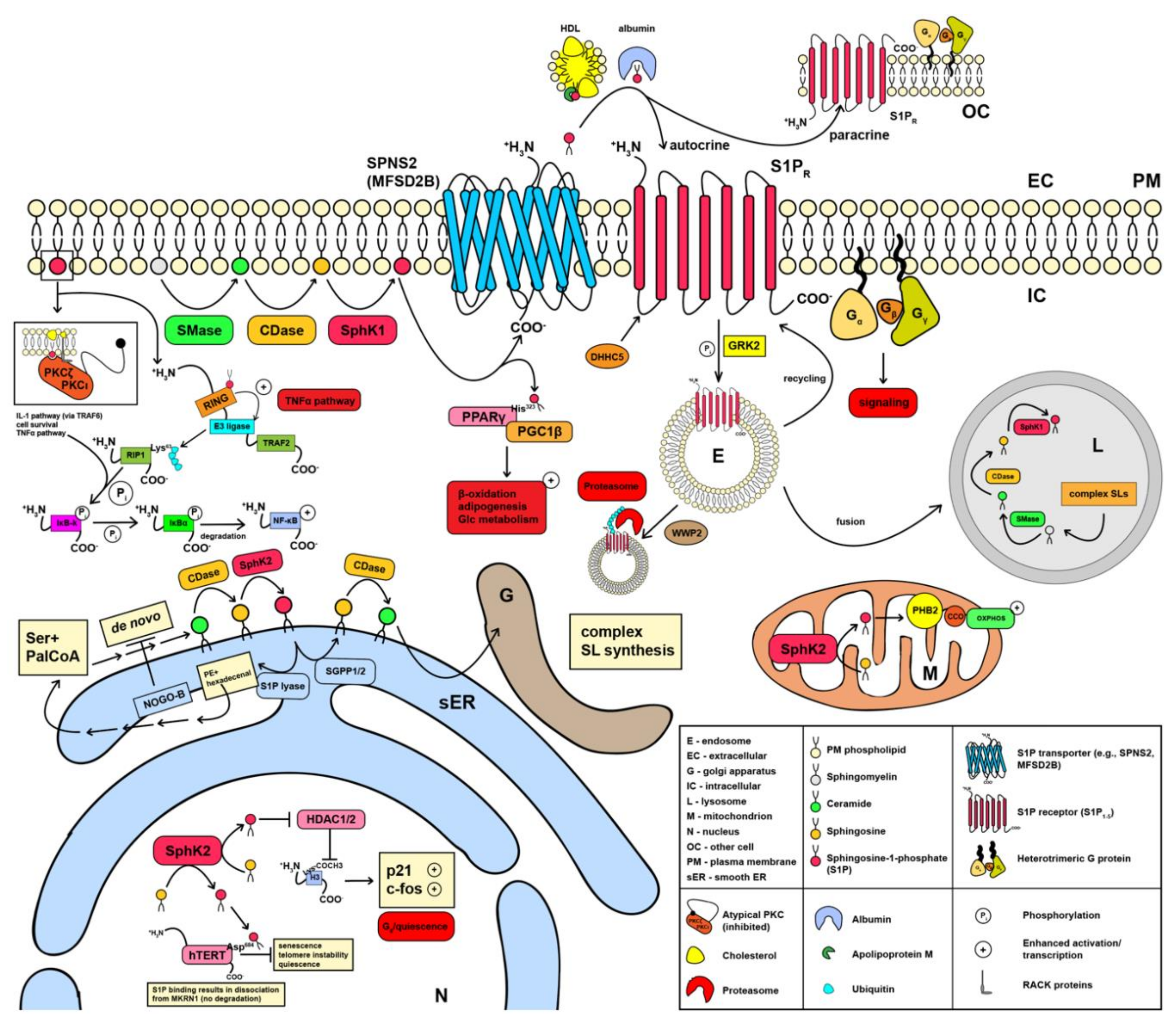{kind=link}
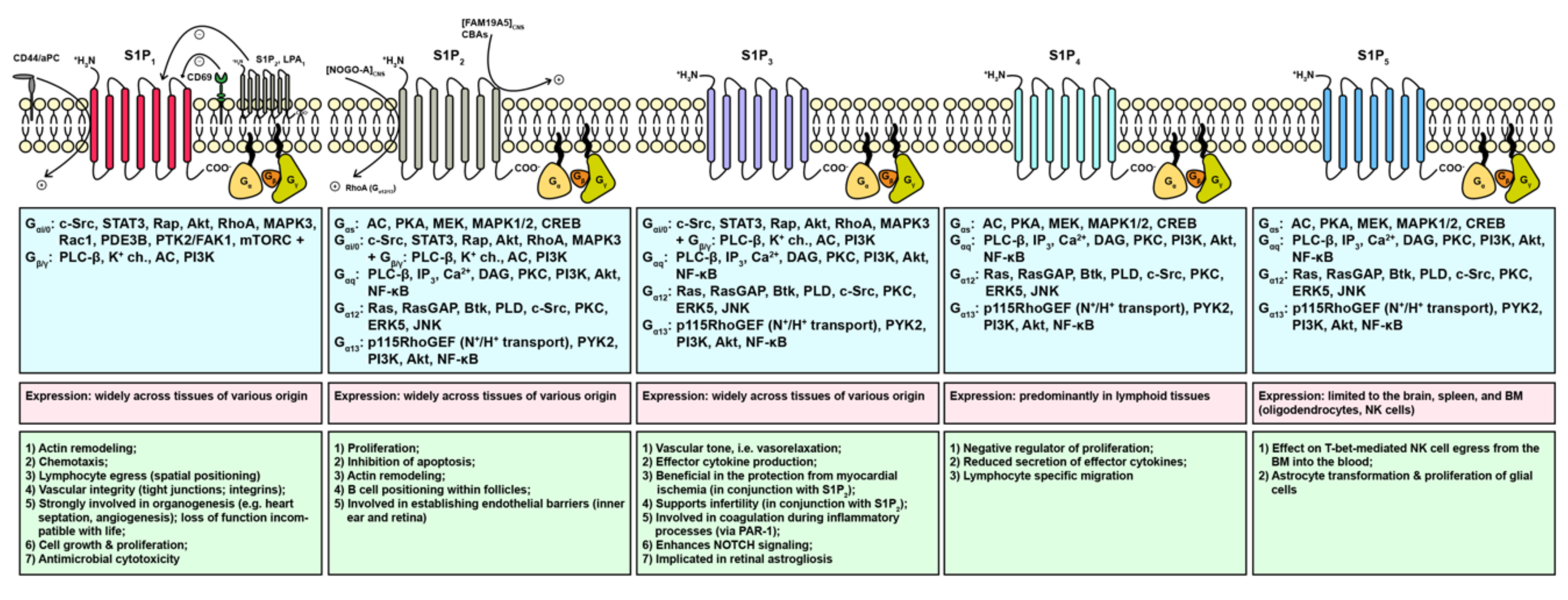{kind=link}
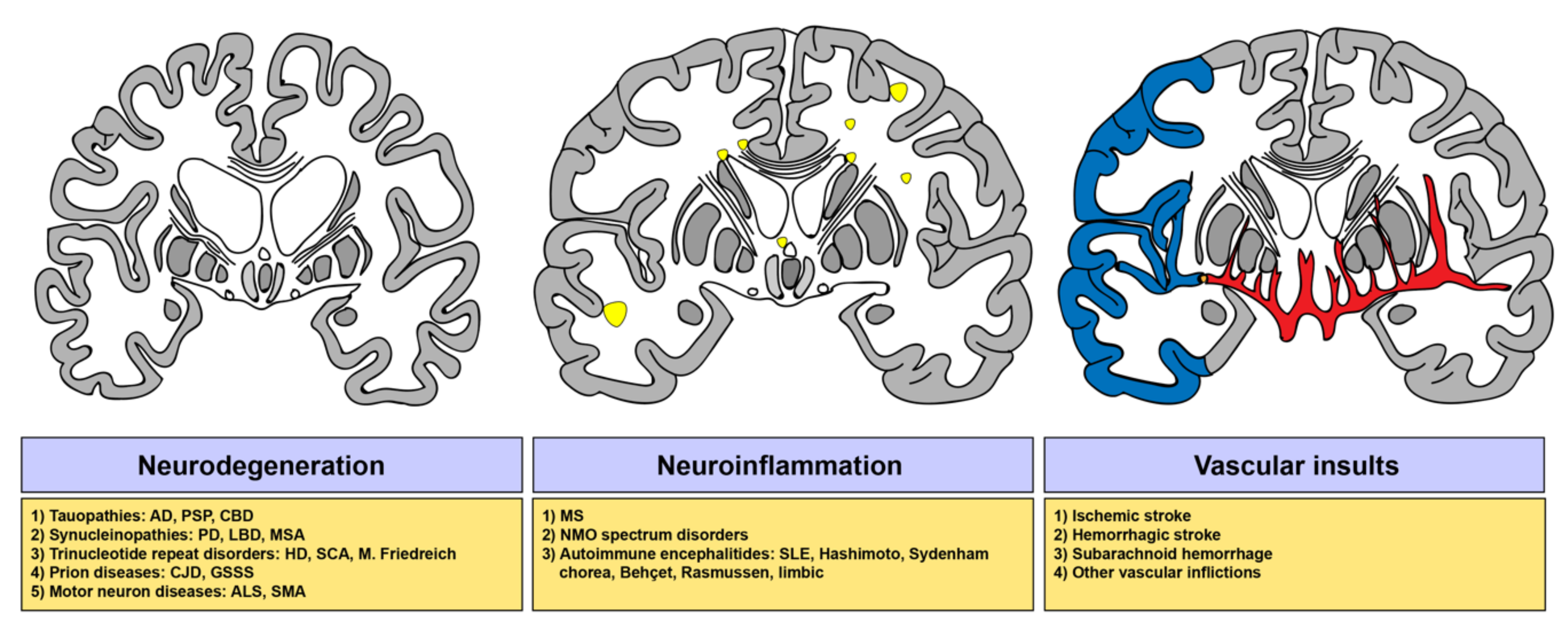{kind=link}
| Target | Compounds (Mechanism of Action) | Indications | ClinicalTrials.gov Identifier | Phase |
|---|---|---|---|---|
| SPHK1/2, PKC inhibitor | Safingol | Solid tumor | NCT01553071 | I |
| Adult solid tumor (unspec.) | NCT00084812 | I (C) | ||
| SPHK2 | ABC294640 (SPHK2 inhibition) | Non-resectable, perihilar cholangiocarcinoma (extra- and intrahepatic) | NCT03377179 | II |
| NCT03414489 | n/a | |||
| Pancreatic cancer, adult solid tumor (unspec.) | NCT01488513 | I (C) | ||
| Multiple myeloma | NCT02757326 | I, II (T) | ||
| S1P | n/a | Bacterial pneumonia | NCT04007328 | II, III |
| Food Allergy, anaphylaxis | NCT01776489 | n/a | ||
| Asthma | NCT04134351 | n/a | ||
| Pneumonia, chronic obstructive pulmonary disease, asthma | NCT03473119 | n/a | ||
| Sonepcizumab [LT1009] (S1P-specific mAb) | Solid tumors | NCT00661414 | I (C) | |
| Neovascular age-related macular degeneration | NCT00767949 | I | ||
| Exudative age-related macular degeneration | NCT01414153 | II (C) | ||
| Pigment epithelial detachment | NCT01334255 | I (T) | ||
| Renal cell carcinoma | NCT01762033 | II (T) | ||
| S1P lyase | LX3305(S1P lyase inhibition) | Rheumatoid arthritis | NCT00847886, NCT01417052 | I (C) |
| NCT00903383 | II (C) | |||
| S1P1 | n/a | Interstitial cystitis | NCT03003845 | n/a |
| Endometriosis | NCT02973854 | n/a | ||
| Vulvodynia | NCT02981433 | n/a | ||
| AKP11 | Atopic dermatitis | ACTRN12617000763347 | II | |
| Rheumatoid arthritis | ACTRN12617001223325 | II | ||
| BMS-986104 | Rheumatoid arthritis (healthy volunteers) | NCT02211469 | II (C) | |
| Cenerimod[ACT-334441](S1P1 agonist) | Systemic lupus erythematosus | NCT02472795 | I, II (C) | |
| Healthy volunteers | NCT04052360 | I (C) | ||
| NCT04255277 | I | |||
| CS-077 (S1P1 agonist) | Multiple sclerosis | NCT00616733 | I (C) | |
| GSK2018682(S1P1 agonist) | Multiple sclerosis (healthy volunteers) | NCT01387217 | I (C) | |
| Relapsing–remitting multiple sclerosis (healthy volunteers) | NCT01466322, NCT01431937 | I (C) | ||
| KRP203 (S1P1 agonist) | Subacute cutaneous lupus erythematosus | NCT01294774 | II (C) | |
| Hematological malignancies | NCT01830010 | I (C) | ||
| Ulcerative colitis | NCT01375179 | II (T) | ||
| Ponesimod [ACT-128800] (S1P1 agonist) | Multiple sclerosis | NCT02425644 | III (C) | |
| NCT03232073 | III | |||
| NCT02907177 | III (T) | |||
| Relapsing–remitting multiple sclerosis | NCT01093326 | II | ||
| NCT01006265 | II (C) | |||
| Plaque psoriasis | NCT00852670, NCT01208090 | II (C) | ||
| Chronic graft versus host disease | NCT02461134 | II (T) | ||
| Healthy volunteers | NCT02136888, NCT02068235, NCT03882255, NCT02223832 | I (C) | ||
| Pharmacokinetics | NCT02126956 | I (C) | ||
| Safety and tolerability | NCT02029482 | I (C) | ||
| S1P1, S1P3, S1P4, S1P5 | Fingolimod [FTY720] (S1PR modulator, S1P1 functional antagonist) | Healthy volunteers | NCT00416845, NCT03757338 | I (C) |
| Multiple sclerosis | NCT00537082, NCT00670449, NCT00333138 | II (C) | ||
| NCT01892722 | III | |||
| NCT00662649, NCT00355134, NCT02939079, NCT00340834 | III (C) | |||
| NCT01647880 | III (T) | |||
| NCT01585298, NCT01333501 | IV (C) | |||
| NCT02232061, NCT01981161, NCT02769689 | IV | |||
| NCT02139696, NCT01592097, NCT01285479, NCT01281657, NCT02225977, NCT02408380, NCT03216915, NCT01811290, NCT02799199, NCT02776072, NCT01442194, NCT02021162, NCT02307877, NCT03243721 | n/a | |||
| Multiple sclerosis (autonomic nervous system dysfunction) | NCT02048072 | IV (C) | ||
| Multiple sclerosis (fatigue) | NCT01490840 | IV (T) | ||
| Multiple sclerosis (cognitive deficits) | NCT02141022 | n/a | ||
| Primary progressive multiple sclerosis | NCT00731692 | III (T) | ||
| Relapsing–remitting multiple sclerosis | NCT00289978, NCT01127750, NCT01201356, NCT01497262, NCT01199861 | III (C) | ||
| NCT01499667, NCT01633112 | III (T) | |||
| NCT01310166, NCT02325440 | IV | |||
| NCT02137707, NCT02307838, NCT01420055, NCT02720107, NCT03257358, NCT02373098, NCT01578330, NCT01623596, NCT01534182, NCT01317004, NCT01498887, NCT01216072, NCT01705236 | IV (C) | |||
| NCT01755871, NCT02342704, NCT03345940 | IV (T) | |||
| NCT01790269, NCT01704183, NCT02335892, NCT02277964 | n/a | |||
| Relapsing–remitting multiple sclerosis (cognition, brain volume loss) | NCT02575365 | IV (T) | ||
| Relapsing–remitting multiple sclerosis (depression) | NCT01436643 | IV (T) | ||
| Acute demyelinating optic neuritis | NCT01757691 | II (T) | ||
| Amyotrophic lateral sclerosis | NCT01786174 | II (C) | ||
| Intracerebral hemorrhage (hypertensive, intraparenchymal), cerebral edema | NCT04088630 | I | ||
| (Acute) stroke, (cerebro-) vascular accident, cerebral stroke | NCT02002390 | II (C) | ||
| Chemotherapy-induced peripheral neuropathy (numbness, pain, tingling) | NCT03943498 | I | ||
| Chronic inflammatory demyelinating polyradiculoneuropathy | NCT01625182 | III (C) | ||
| Breast carcinoma | NCT03941743 | I | ||
| Glioblastoma, anaplastic astrocytoma | NCT02490930 | I (C) | ||
| Coronavirus disease (COVID-19) | NCT04280588 | II | ||
| Asthma | NCT00785083 | II (C) | ||
| Rett syndrome | NCT02061137 | II (C) | ||
| Schizophrenia | NCT01779700 | II (C) | ||
| Renal insufficiency | NCT00731523 | I (C) | ||
| Kidney transplantation | NCT00239902, NCT00239798 | II (C) | ||
| NCT00099736, NCT00239876, NCT00239811, NCT00099801, NCT00099749, NCT00239863, NCT00239785, NCT00098735 | III (C) | |||
| S1P1, S1P5 | Ceralifimod [ONO-4641] (S1P1,5 agonist) | Multiple sclerosis | NCT01081782 | II (C) |
| NCT01226745 | II (T) | |||
| Ozanimod [RPC1063] (S1P1,5 agonist) | Multiple sclerosis | NCT02797015 | I (C) | |
| NCT02576717, NCT04140305 | III | |||
| NCT02294058 | III (C) | |||
| Relapsing–remitting multiple sclerosis | NCT01628393, NCT02047734 | III (C) | ||
| Ulcerative colitis | NCT01647516 | II | ||
| NCT02435992, NCT02531126, NCT03915769 | III | |||
| Crohn’s disease | NCT02531113 | II (C) | ||
| NCT03467958, NCT03464097, NCT03440372, NCT03440385 | III | |||
| Healthy volunteers | NCT02994381, NCT03694119, NCT03644576, NCT03624959, NCT03665610 | I (C) | ||
| NCT04149678, NCT04211558 | I | |||
| Siponimod [BAF312] (S1P1,5 modulator) | Healthy volunteers | NCT00422175 | I (C) | |
| Multiple sclerosis | NCT03623243 | III | ||
| Relapsing-remitting multiple sclerosis | NCT01185821, NCT00879658 | II (C) | ||
| Secondary progressive multiple sclerosis | NCT01665144 | III | ||
| NCT02330965 | n/a | |||
| Polymyositis (, dermato-myositis) | NCT01801917, NCT01148810, NCT02029274 | II (T) | ||
| Hepatic impairment | NCT01565902 | I (C) | ||
| Hemorrhagic stroke, intracerebral hemorrhage (ICH) | NCT03338998 | II (S) | ||
| Renal impairment | NCT01904214 | I (C) | ||
| S1P1, S1P5, (S1P4) | Amiselimod [MT-1303] (S1PR modulator, S1P1 functional antagonist) | Relapsing–remitting multiple sclerosis | NCT02193217, NCT02310048, NCT02293967 | I (C) |
| NCT01890655, NCT01742052 | II (C) | |||
| Crohn’s disease | NCT02148185 | I (C) | ||
| NCT02389790, NCT02378688 | II (C) | |||
| Systemic lupus erythematosus | NCT02307643 | I (C) | ||
| Plaque psoriasis | NCT01987843 | II (C) | ||
| Inflammatory bowel disease | NCT01666327 | I (C) | ||
| Etrasimod [APD334] | Primary biliary cholangitis | NCT03155932 | II (T) | |
| Inflammatory bowel disease(extra-int. skin manifestations) | NCT03139032 | II (T) | ||
| Ulcerative colitis | NCT02447302, NCT02536404 | II (C) | ||
| NCT03950232, NCT03945188, NCT03996369, NCT04176588 | III | |||
| Pyoderma gangrenosum | NCT03072953 | II (T) | ||
| Crohn’s disease | NCT04173273 | II | ||
| Atopic dermatitis | NCT04162769 | II |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucaciu, A.; Brunkhorst, R.; Pfeilschifter, J.M.; Pfeilschifter, W.; Subburayalu, J. The S1P–S1PR Axis in Neurological Disorders—Insights into Current and Future Therapeutic Perspectives. Cells 2020, 9, 1515. https://doi.org/10.3390/cells9061515
Lucaciu A, Brunkhorst R, Pfeilschifter JM, Pfeilschifter W, Subburayalu J. The S1P–S1PR Axis in Neurological Disorders—Insights into Current and Future Therapeutic Perspectives. Cells. 2020; 9(6):1515. https://doi.org/10.3390/cells9061515
Chicago/Turabian StyleLucaciu, Alexandra, Robert Brunkhorst, Josef M. Pfeilschifter, Waltraud Pfeilschifter, and Julien Subburayalu. 2020. "The S1P–S1PR Axis in Neurological Disorders—Insights into Current and Future Therapeutic Perspectives" Cells 9, no. 6: 1515. https://doi.org/10.3390/cells9061515
APA StyleLucaciu, A., Brunkhorst, R., Pfeilschifter, J. M., Pfeilschifter, W., & Subburayalu, J. (2020). The S1P–S1PR Axis in Neurological Disorders—Insights into Current and Future Therapeutic Perspectives. Cells, 9(6), 1515. https://doi.org/10.3390/cells9061515