Lessons from the Endoplasmic Reticulum Ca2+ Transporters—A Cancer Connection


{kind=link}
{kind=link}
Abstract
:1. Introduction
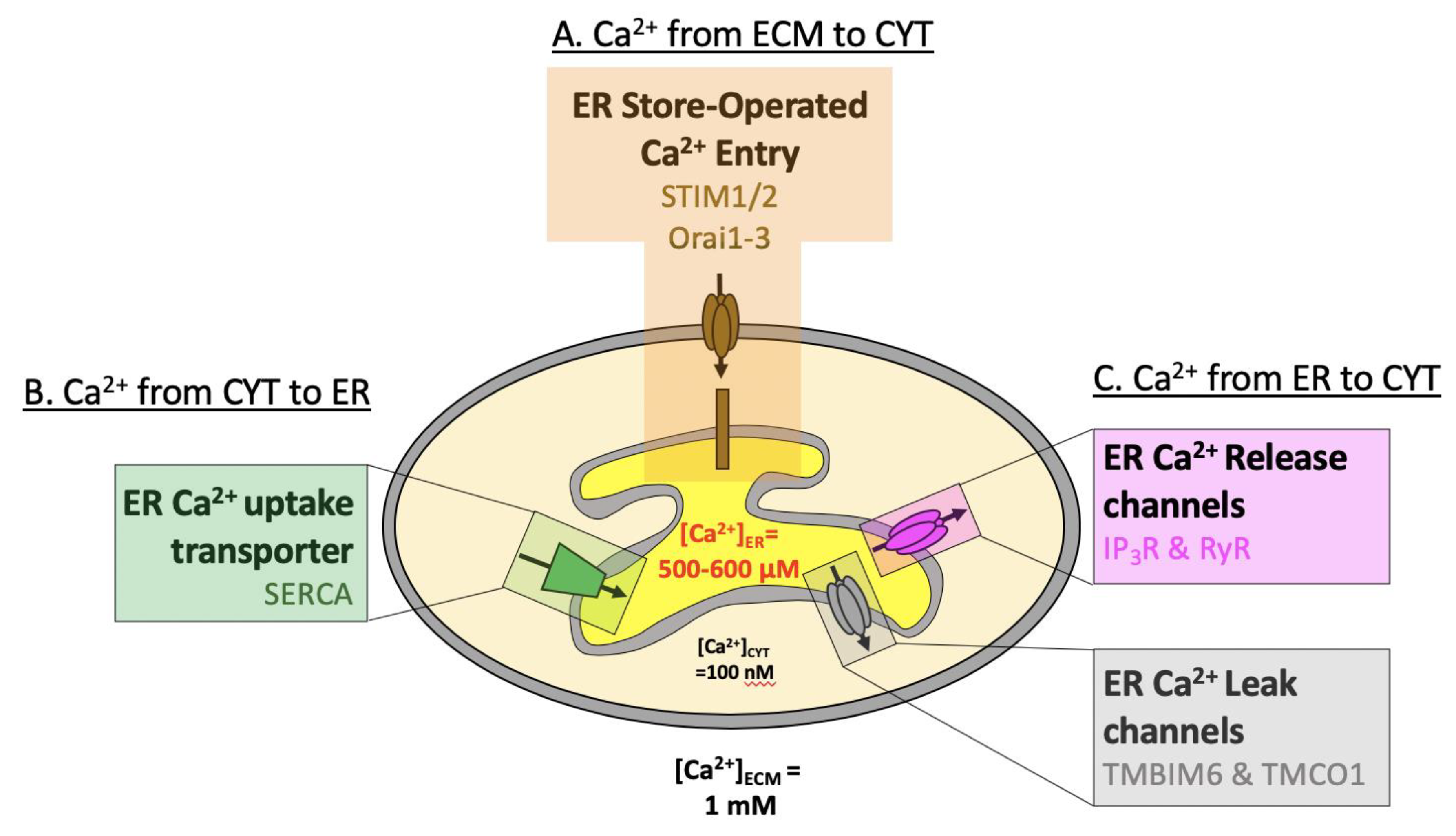
2. ER Topography and Ca2+ Handling
3. ER Ca2+-Releasing Channels
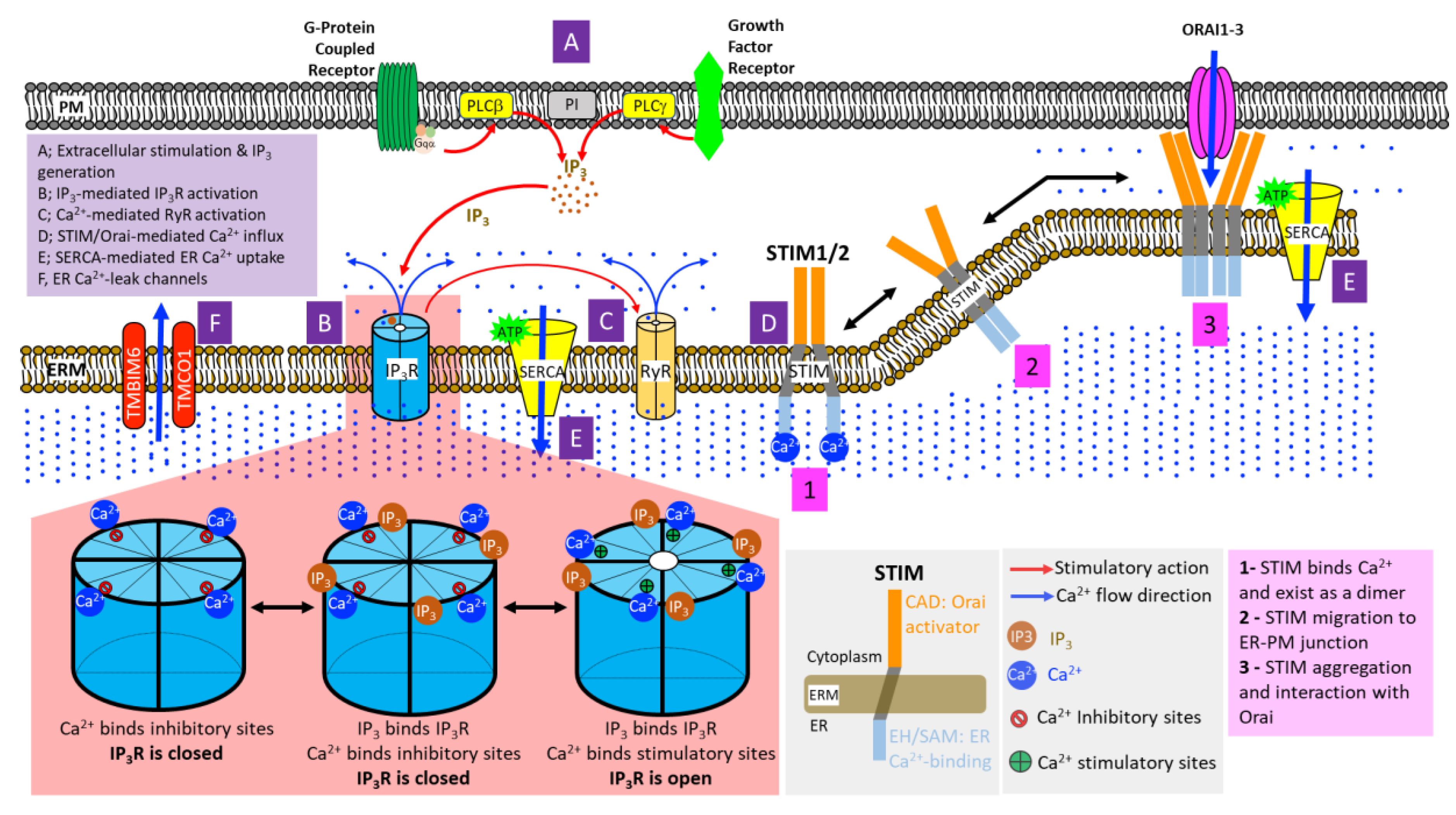
3.1. Inositol 1,4,5-Trisphosphate Receptors (IP3Rs)
3.2. Ryanodine Receptors (RyRs)
4. ER Ca2+ Replenishment
4.1. STIM-Orai
4.2. SERCAs and ER Ca2+-Refilling
5. ER Ca2+ Transporters and Cancer Pathophysiology
5.1. IP3Rs in Cancer
5.2. RyRs in Cancer
5.3. STIM-Orai Channels in Cancer
5.4. SERCAs in Cancer
6. Targeting ER Ca2+ Signaling in Anti-Cancer Therapy
7. Closing Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sjöstrand, F.S. The endoplasmic reticulum. In Cytology and Cell Physiology, 3rd ed.; Academic Press: London, UK, 1964; pp. 311–375. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The endoplasmic reticulum. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Filadi, R.; Zampese, E.; Pozzan, T.; Pizzo, P.; Fasolato, C. Endoplasmic reticulum-mitochondria connections, calcium cross-talk and cell fate: A closer inspection. In Endoplasmic Reticulum Stress in Health and Disease; Springer Netherlands: Dordrecht, The Netherlands, 2012; pp. 75–106. [Google Scholar]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Wang, X.; Patel, S.; Clapham, D.E. Insights into the early evolution of animal calcium signaling machinery: A unicellular point of view. Cell Calcium 2015, 57, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Carafoli, E.; Krebs, J. Why calcium? How calcium became the best communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stefan, M.I.; Le Novère, N. Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PLoS ONE 2012, 7, e43810. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bong, A.H.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Sun, S.; Hu, J. Molecular basis for sculpting the endoplasmic reticulum membrane. Int. J. Biochem. Cell Biol. 2012, 44, 1436–1443. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The transport of molecules between the nucleus and the cytosol. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Protein glycosylation in the ER and Golgi complex. In Molecular Cell Biology, 4th ed.; WH Freeman: New York, NY, USA, 2000. [Google Scholar]
- Fewell, S.W.; Brodsky, J.L. Entry into the endoplasmic reticulum: Protein translocation, folding and quality control. In Trafficking Inside Cells; Springer: New York, NY, USA, 2009; pp. 119–142. [Google Scholar]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar]
- Kang, M.; Othmer, H.G. The variety of cytosolic calcium responses and possible roles of PLC and PKC. Phys. Biol. 2007, 4, 325. [Google Scholar] [CrossRef] [PubMed]
- Laude, A.J.; Simpson, A.W. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C.; Bootman, M.D.; Scott, J.D. Second messengers. Cold Spring Harb. Perspect. Biol. 2016, 8, a005926. [Google Scholar] [CrossRef] [PubMed]
- Serysheva, I.I. Toward a high-resolution structure of IP3R channel. Cell Calcium 2014, 56, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serysheva, I.I.; Baker, M.R.; Fan, G. Structural insights into IP 3 R function. In Membrane Dynamics and Calcium Signaling; Springer International Publishing: Cham, Switzerland, 2017; pp. 121–147. [Google Scholar]
- Sharp, A.H.; Nucifora, F.C., Jr.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugoand, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1, 4, 5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1, 4, 5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef] [Green Version]
- Hirata, K.; Pusl, T.; O’Neill, A.F.; Dranoff, J.A.; Nathanson, M.H. The type II inositol 1, 4, 5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology 2002, 122, 1088–1100. [Google Scholar] [CrossRef]
- Iwai, M.; Michikawa, T.; Bosanac, I.; Ikura, M.; Mikoshiba, K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1, 4, 5-trisphosphate receptors. J. Biol. Chem. 2007, 282, 12755–12764. [Google Scholar] [CrossRef] [Green Version]
- Fujino, I.; Yamada, N.; Miyawaki, A.; Hasegawa, M.; Furuichi, T.; Mikoshiba, K. Differential expression of type 2 and type 3 inositol 1, 4, 5-trisphosphate receptor mRNAs in various mouse tissues: In situ hybridization study. Cell Tissue Res. 1995, 280, 201–210. [Google Scholar]
- De Young, G.W.; Keizer, J. A single-pool inositol 1, 4, 5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. USA 1992, 89, 9895–9899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Othmer, H.G.; Tang, Y. Oscillations and waves in a model of InsP 3-controlled calcium dynamics. In Experimental and Theoretical Advances in Biological Pattern Formation; Springer: Boston, MA, USA, 1993; pp. 277–300. [Google Scholar]
- Tang, Y.; Stephenson, J.L.; Othmer, H.G. Simplification and analysis of models of calcium dynamics based on IP3-sensitive calcium channel kinetics. Biophys. J. 1996, 70, 246–263. [Google Scholar] [CrossRef] [Green Version]
- Alzayady, K.J.; Wang, L.; Chandrasekhar, R.; Wagner, L.E.; Van Petegem, F.; Yule, D.I. Defining the stoichiometry of inositol 1, 4, 5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 2016, 9, ra35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, K.; Miyatake, H.; Terauchi, A.; Mikoshiba, K. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 4661–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.W.; Tovey, S.C. IP3 receptors: Toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; McBride, S.; Mak, D.-O.D.; Vardi, N.; Palczewski, K.; Haeseleer, F.; Foskett, J.K. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. USA 2002, 99, 7711–7716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca 2+ signals initiate at immobile IP 3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Structure and function of IP3 receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic reticulum–mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP 3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca2+ to lysosomes. Cell Rep. 2018, 25, 3180–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.Y.; Jha, A.; Ahuja, M.; Muallem, S. Ca2+ influx at the ER/PM junctions. Cell Calcium 2017, 63, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Yang, Y.D.; Lee, J.; Lee, B.; Kim, T.; Jang, Y.; Back, S.K.; Na, H.S.; Harfe, B.D.; Wang, F.; et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat. Neurosci. 2012, 15, 1015. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.J.; Bootman, M.D.; Roderick, H.L. Cell signalling: IP3 receptors channel calcium into cell death. Curr. Biol. 2004, 14, R933–R935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, G.; Cruz, P.; Lovy, A.; Cárdenas, C. Endoplasmic reticulum–mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: A novel potential target. Front. Oncol. 2017, 7, 199. [Google Scholar] [CrossRef] [Green Version]
- López-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins (1, 4, 5) P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzio, J.P.; Gray, S.R.; Bright, N.A. Endosome–lysosome fusion. In Proceedings of the Lysosomes in Health and Disease, Charles Darwin House, London, UK, 13–14 May 2010; pp. 1413–1416. [Google Scholar]
- Lock, J.T.; Alzayady, K.J.; Yule, D.I.; Parker, I. All three IP3 receptor isoforms generate Ca2+ puffs that display similar characteristics. Sci. Signal. 2018, 11, eaau0344. [Google Scholar]
- Alzayady, K.J.; Wojcikiewicz, R.J. The role of Ca2+ in triggering inositol 1, 4, 5-trisphosphate receptor ubiquitination. Biochem. J. 2005, 392, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Wojcikiewicz, R.J. Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol. Sci. 2004, 25, 35–41. [Google Scholar] [CrossRef]
- Vervloessem, T.; Yule, D.I.; Bultynck, G.; Parys, J.B. The type 2 inositol 1, 4, 5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim. Biophys. Acta 2015, 1853, 1992–2005. [Google Scholar] [CrossRef] [Green Version]
- Mak, D.-O.D.; McBride, S.M.; Petrenko, N.B.; Foskett, J.K. Novel regulation of calcium inhibition of the inositol 1, 4, 5-trisphosphate receptor calcium-release channel. J. Gen. Physiol. 2003, 122, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyakawa, T.; Maeda, A.; Yamazawa, T.; Hirose, K.; Kurosaki, T.; Iino, M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999, 18, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Hegg, C.C.; Jia, C.; Chick, W.S.; Restrepo, D.; Hansen, A. Microvillous cells expressing IP3 receptor type 3 in the olfactory epithelium of mice. Eur. J. Neurosci. 2010, 32, 1632–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, S.K.; Arendshorst, W.J. Voltage-gated Ca2+ entry and ryanodine receptor Ca2+-induced Ca2+ release in preglomerular arterioles. Am. J. Physiol. Renal Physiol. 2007, 292, F1568–F1572. [Google Scholar] [CrossRef] [Green Version]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef] [Green Version]
- Meissner, G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 2004, 35, 621–628. [Google Scholar] [CrossRef]
- Balshaw, D.M.; Xu, L.; Yamaguchi, N.; Pasek, D.A.; Meissner, G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 2001, 276, 20144–20153. [Google Scholar] [CrossRef] [Green Version]
- Camors, E.; Valdivia, H.H. CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Front. Pharmacol. 2014, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Zima, A.V.; Copello, J.A.; Blatter, L.A. Differential modulation of cardiac and skeletal muscle ryanodine receptors by NADH. FEBS Lett. 2003, 547, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Laver, D.; Baynes, T.; Dulhunty, A. Magnesium inhibition of ryanodine-receptor calcium channels: Evidence for two independent mechanisms. J. Membr. Biol. 1997, 156, 213–229. [Google Scholar] [CrossRef]
- Yao, Y.; Choi, J.; Parker, I. Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J. Physiol. 1995, 482, 533–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Miyashita, Y.; Kasai, H. Micromolar and submicromolar Ca2+ spikes regulating distinct cellular functions in pancreatic acinar cells. EMBO J. 1997, 16, 242–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang-Trong, T.M.; Ullah, A.; Jafri, M.S. Calcium sparks in the heart: Dynamics and regulation. Res. Rep. Biol. 2015, 6, 203. [Google Scholar]
- Lipp, P.; Thomas, D.; Berridge, M.J.; Bootman, M.D. Nuclear calcium signalling by individual cytoplasmic calcium puffs. EMBO J. 1997, 16, 7166–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foskett, J.K.; White, C.; Cheung, K.-H.; Mak, D.-O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Rondé, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, G.D.; Swaminathan, D.; Parker, I. The probability of triggering calcium puffs is linearly related to the number of inositol trisphosphate receptors in a cluster. Biophys. J. 2012, 102, 1826–1836. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Prakriya, M. The molecular physiology of CRAC channels. Immunol. Rev. 2009, 231, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.; Kim, M.L.; Do Heo, W.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Nwokonko, R.M.; Cai, X.; Loktionova, N.A.; Abdulqadir, R.; Xin, P.; Niemeyer, B.A.; Wang, Y.; Trebak, M.; Gill, D.L. Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3398–E3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphaël, M.; Lehen’kyi, V.y.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Abeele, F.V.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elaib, Z.; Saller, F.; Bobe, R. The calcium entry-calcium refilling coupling. In Calcium Entry Pathways in Non-Excitable Cells; Springer International Publishing: Cham, Switerland, 2016; pp. 333–352. [Google Scholar]
- Møller, J.V.; Olesen, C.; Winther, A.-M.L.; Nissen, P. The sarcoplasmic Ca 2+-ATPase: Design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 2010, 43, 501–566. [Google Scholar] [CrossRef]
- Gélébart, P.; Martin, V.; Enouf, J.; Papp, B. Identification of a new SERCA2 splice variant regulated during monocytic differentiation. Biochem. Biophys. Res. Commun. 2003, 303, 676–684. [Google Scholar] [CrossRef]
- Altshuler, I.; Vaillant, J.J.; Xu, S.; Cristescu, M.E. The evolutionary history of sarco (endo) plasmic calcium ATPase (SERCA). PLoS ONE 2012, 7, e52617. [Google Scholar] [CrossRef] [Green Version]
- Dally, S.; Corvazier, E.; Bredoux, R.; Bobe, R.; Enouf, J. Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J. Mol. Cell. Cardiol. 2010, 48, 633–644. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Bobe, R.; Bredoux, R.; Corvazier, E.; Lacabaratz-Porret, C.; Martin, V.; Kovacs, T.; Enouf, J. How many Ca2+ ATPase isoforms are expressed in a cell type? A growing family of membrane proteins illustrated by studies in platelets. Platelets 2005, 16, 133–150. [Google Scholar] [CrossRef]
- Corvazier, E.; Bredoux, R.; Kovács, T.; Enouf, J. Expression of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) 3 proteins in two major conformational states in native human cell membranes. Biochim. Et Biophys. Acta 2009, 1788, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Sakuta, N.; Watanabe, S.; Zhang, Y.; Yoshikaie, K.; Tanaka, Y.; Ushioda, R.; Kato, Y.; Takagi, J.; Tsukazaki, T.; et al. Structural Basis of Sarco/Endoplasmic Reticulum Ca2+-ATPase 2b Regulation via Transmembrane Helix Interplay. Cell Rep. 2019, 27, 1221–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecaetsbeek, I.; Trekels, M.; De Maeyer, M.; Ceulemans, H.; Lescrinier, E.; Raeymaekers, L.; Wuytack, F.; Vangheluwe, P. Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc. Natl. Acad. Sci. USA 2009, 106, 18533–18538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekera, P.C.; Kargacin, M.E.; Deans, J.P.; Lytton, J. Determination of apparent calcium affinity for endogenously expressed human sarco (endo) plasmic reticulum calcium-ATPase isoform SERCA3. Am. J. Physiol. Cell Physiol. 2009, 296, C1105–C1114. [Google Scholar] [CrossRef] [Green Version]
- Martin, V.; Bredoux, R.; Corvazier, E.; Van Gorp, R.; Kovàcs, T.; Gélébart, P.; Enouf, J. Three novel sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 3 isoforms expression, regulation, and function of the members of the SERCA3 family. J. Biol. Chem. 2002, 277, 24442–24452. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.R.; Vangheluwe, P. Primary active Ca2+ transport systems in health and disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035113. [Google Scholar] [CrossRef]
- Bilmen, J.G.; Khan, S.Z.; Javed, M.u.H.; Michelangeli, F. Inhibition of the SERCA Ca2+ pumps by curcumin: Curcumin putatively stabilizes the interaction between the nucleotide-binding and phosphorylation domains in the absence of ATP. Eur. J. Biochem. 2001, 268, 6318–6327. [Google Scholar] [CrossRef]
- Asahi, M.; Green, N.M.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Phospholamban domain IB forms an interaction site with the loop between transmembrane helices M6 and M7 of sarco (endo) plasmic reticulum Ca2+ ATPases. Proc. Natl. Acad. Sci. USA 2001, 98, 10061–10066. [Google Scholar] [CrossRef] [Green Version]
- Dicke, A.A.; Gopinath, T.; Vostrikov, V.V.; Veglia, G. The Effects of Sarcolipin Phosphorylation on SERCA Regulation. Biophys. J. 2016, 110, 395a. [Google Scholar] [CrossRef]
- MacLENNAN, D.H.; Toyofuku, T.; Kimura, Y. Sites of regulatory interaction between calcium ATPases and phospholamban. In Alterations of Excitation-Contraction Coupling in the Failing Human Heart; Steinkopff-Verlag: Heidelberg, Germany, 1998; pp. 17–24. [Google Scholar]
- Lisak, D.A.; Schacht, T.; Enders, V.; Habicht, J.; Kiviluoto, S.; Schneider, J.; Henke, N.; Bultynck, G.; Methner, A.; Nickel, N. The transmembrane Bax inhibitor motif (TMBIM) containing protein family: Tissue expression, intracellular localization and effects on the ER CA2+-filling state. Biochim. Biophys. Acta 2015, 1853, 2104–2114. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q. TMBIM-mediated Ca2+ homeostasis and cell death. Biochim. Biophys. Acta 2017, 1864, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Kiviluoto, S.; Methner, A. Bax inhibitor-1 is likely a pH-sensitive calcium leak channel, not a H+/Ca2+ exchanger. Sci. Signal. 2014, 7, pe22. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Bruni, R.; Kloss, B.; Assur, Z.; Kloppmann, E.; Rost, B.; Hendrickson, W.A.; Liu, Q. Structural basis for a pH-sensitive calcium leak across membranes. Science 2014, 344, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Xu, M.; Chang, Y.; Luyten, T.; Seitaj, B.; Liu, W.; Zhu, P.; Bultynck, G.; Shi, L.; Quick, M.; et al. Ion and pH Sensitivity of a TMBIM Ca2+ Channel. Structure 2019, 27, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Kiviluoto, S.; Henke, N.; Ivanova, H.; Schneider, L.; Rybalchenko, V.; Luyten, T.; Nuyts, K.; De Borggraeve, W.; Bezprozvanny, I.; et al. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. J. Biol. Chem. 2012, 287, 2544–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.-C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.; Kosta, A.; Luciani, M.-F.; Golstein, P. The inositol 1, 4, 5-trisphosphate receptor is required to signal autophagic cell death. Mol. Biol. Cell 2008, 19, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Huang, J.; Liu, W.; Kou, X.; Tang, H.; Wang, H.; Yu, X.; Gao, S.; Ouyang, K.; Yang, H.-T. IP3R-mediated Ca2+ signals govern hematopoietic and cardiac divergence of Flk1+ cells via the calcineurin–NFATc3–Etv2 pathway. J. Mol. Cell Biol. 2017, 9, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.-P.; Aromolaran, A.S.; Bultynck, G.; Zhong, F.; Li, X.; McColl, K.; Matsuyama, S.; Herlitze, S.; Roderick, H.L.; Bootman, M.D.; et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol. Cell 2008, 31, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, Y.-P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [Green Version]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.; De Smedt, H.; et al. Selective regulation of IP 3-receptor-mediated Ca 2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Wagner, L.E.; Tanimura, A.; Vandermarliere, E.; Luyten, T.; Welkenhuyzen, K.; Alzayady, K.J.; Wang, L.; Hamada, K.; Mikoshiba, K.; et al. Mikoshiba, K. Bcl-2 and IP 3 compete for the ligand-binding domain of IP 3 Rs modulating Ca 2+ signaling output. Cell. Mol. Life Sci. 2019, 76, 3843–3859. [Google Scholar] [CrossRef]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The endoplasmic reticulum gateway to apoptosis by Bcl-X L modulation of the InsP 3 R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1, 4, 5-trisphosphate receptor channel isoform gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1, 4, 5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.-P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2–IP3 receptor interaction. Blood J. Am. Soc. Hematol. 2011, 117, 2924–2934. [Google Scholar] [CrossRef] [Green Version]
- Lavik, A.R.; Zhong, F.; Chang, M.-J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 27388. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, E.; McColl, K.; Zhong, F.; Wildey, G.; Dowlati, A.; Distelhorst, C. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1, 4, 5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015, 6, e2034. [Google Scholar] [CrossRef] [Green Version]
- Distelhorst, C.W. Targeting Bcl-2-IP3 receptor interaction to treat cancer: A novel approach inspired by nearly a century treating cancer with adrenal corticosteroid hormones. Biochim. Biophys. Acta 2018, 1865, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Distelhorst, C.W.; Bootman, M.D. Creating a new cancer therapeutic agent by targeting the interaction between Bcl-2 and IP3 receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3-and Ca 2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca 2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierro, C.; Cook, S.J.; Foets, T.C.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Sakakura, C.; Miyagawa, K.; Fukuda, K.; Shimomura, K.; Takemura, M.; Takagi, T.; Kin, S.; Nakase, Y.; Fujiyama, J.; Mikoshiba, K.; et al. Possible involvement of inositol 1, 4, 5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Gan Kagaku Ryoho. Cancer Chemother. 2003, 30, 1784–1787. [Google Scholar]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Inhibition of the Ca2+ release channel, IP3R subtype 3 by caffeine slows glioblastoma invasion and migration and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1, 4, 5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef] [Green Version]
- Mound, A.; Vautrin-Glabik, A.; Foulon, A.; Botia, B.; Hague, F.; Parys, J.B.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. Downregulation of type 3 inositol (1, 4, 5)-trisphosphate receptor decreases breast cancer cell migration through an oscillatory Ca2+ signal. Oncotarget 2017, 8, 72324. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1, 4, 5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Vautrin-Glabik, A.; Botia, B.; Kischel, P.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. IP3R3 silencing induced actin cytoskeletal reorganization through ARHGAP18/RhoA/mDia1/FAK pathway in breast cancer cell lines. Biochim. Biophys. Acta 2018, 1865, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Shibao, K.; Fiedler, M.J.; Nagata, J.; Minagawa, N.; Hirata, K.; Nakayama, Y.; Iwakiri, Y.; Nathanson, M.H.; Yamaguchi, K. The type III inositol 1, 4, 5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010, 48, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, B.; Hudecova, S.; Lencesova, L.; Babula, P.; Rezuchova, I.; Penesova, A.; Grman, M.; Moravcik, R.; Zeman, M.; Krizanova, O. Melatonin-induced changes in cytosolic calcium might be responsible for apoptosis induction in tumour cells. Cell. Physiol. Biochem. 2017, 44, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Hudecova, S.; Markova, J.; Simko, V.; Csaderova, L.; Stracina, T.; Sirova, M.; Fojtu, M.; Svastova, E.; Gronesova, P.; Pastorek, M.; et al. Sulforaphane-induced apoptosis involves the type 1 IP3 receptor. Oncotarget 2016, 7, 61403–61418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunoda, T.; Koga, H.; Yokomizo, A.; Tatsugami, K.; Eto, M.; Inokuchi, J.; Hirata, A.; Masuda, K.; Okumura, K.; Naito, S. Inositol 1, 4, 5-trisphosphate (IP 3) receptor type1 (IP 3 R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 2005, 24, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Shui, B.; Zhao, W.; Liu, H.; Li, W.; Lee, J.C.; Doran, R.; Lee, F.K.; Sun, T.; Shen, Q.S.; et al. Central role of IP 3 R2-mediated Ca 2+ oscillation in self-renewal of liver cancer stem cells elucidated by high-signal ER sensor. Cell Death Dis. 2019, 10, 396. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Jin, M.; Chen, Y.-X.; Wang, J.; Zhai, K.; Chang, Y.; Yuan, Q.; Yao, K.-T.; Ji, G. ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging 2016, 8, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Bittremieux, M.; La Rovere, R.M.; Akl, H.; Martines, C.; Welkenhuyzen, K.; Dubron, K.; Baes, M.; Janssens, A.; Vandenberghe, P.; Laurenti, L.; et al. Constitutive IP 3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP 3 receptor disruptor BIRD-2. Cell Death Differ. 2019, 26, 531–547. [Google Scholar] [CrossRef]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgó, J.; Distelhorst, C.; Missiaen, L.; Mikoshiba, K.; et al. IP 3 R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP 3 R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Witherspoon, J.W.; Meilleur, K.G. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol. Commun. 2016, 4, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ather, S.; Respress, J.L.; Li, N.; Wehrens, X.H. Alterations in ryanodine receptors and related proteins in heart failure. Biochim. Biophys. Acta 2013, 1832, 2425–2431. [Google Scholar] [CrossRef] [Green Version]
- Murayama, T.; Ogawa, Y. Properties of Ryr3 ryanodine receptor isoform in mammalian brain. J. Biol. Chem. 1996, 271, 5079–5084. [Google Scholar] [PubMed] [Green Version]
- Abdul, M.; Ramlal, S.; Hoosein, N. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol. Oncol. Res. 2008, 14, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhang, D.; Chen, J.; He, G.; Gao, L. Low expression of ryanodine receptor 2 is associated with poor prognosis in thyroid carcinoma. Oncol. Lett. 2019, 18, 3605–3612. [Google Scholar] [CrossRef] [Green Version]
- Deli, T.; Varga, N.; Ádám, A.; Kenessey, I.; Rásó, E.; Puskás, L.G.; Tóvári, J.; Fodor, J.; Fehér, M.; Szigeti, G.P.; et al. Functional genomics of calcium channels in human melanoma cells. Int. J. Cancer 2007, 121, 55–65. [Google Scholar] [CrossRef]
- Bennett, D.L.; Cheek, T.R.; Berridge, M.J.; De Smedt, H.; Parys, J.B.; Missiaen, L.; Bootman, M.D. Expression and function of ryanodine receptors in nonexcitable cells. J. Biol. Chem. 1996, 271, 6356–6362. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liang, H.; Lin, C.; Li, F.; Xie, G.; Qiao, S.; Shi, X.; Deng, J.; Zhao, X.; Wu, K.; et al. Molecular subtyping and prognostic assessment based on tumor mutation burden in patients with lung adenocarcinomas. Int. J. Mol. Sci. 2019, 20, 4251. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, Y.; Song, F.; Zheng, H.; Hu, L.; Lu, H.; Liu, P.; Hao, X.; Zhang, W.; Chen, K. Functional SNP in the microRNA-367 binding site in the 3′ UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc. Natl. Acad. Sci. USA 2011, 108, 13653–13658. [Google Scholar] [CrossRef] [Green Version]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; Dias, I.R.D.S.R.; Javed, M.-U.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine induces autophagy-dependent cell death in apoptosis-resistant cancers via ryanodine receptor and Ca 2+-dependent mechanism. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca2+ release stimulates breast cancer stem cell enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Mackrill, J.J. Ryanodine receptor calcium channels and their partners as drug targets. Biochem. Pharmacol. 2010, 79, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuderland, D.; Seger, R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-Y.; Sun, J.; Huang, M.-Y.; Wang, Y.-S.; Hou, M.-F.; Sun, Y.; He, H.; Krishna, N.; Chiu, S.-J.; Lin, S.; et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 2015, 34, 4358–4367. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Chiu, W.-T.; Chen, Y.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.-C.; Kim, M.; et al. STIM1-and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Okeke, E.; Parker, T.; Dingsdale, H.; Concannon, M.; Awais, M.; Voronina, S.; Molgó, J.; Begg, M.; Metcalf, D.; Knight, A.E.; et al. Epithelial–mesenchymal transition, IP3 receptors and ER–PM junctions: Translocation of Ca2+ signalling complexes and regulation of migration. Biochem. J. 2016, 473, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [Green Version]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuttleworth, T. What drives calcium entry during [Ca2+] ioscillations?–challenging the capacitative model. Cell Calcium 1999, 25, 237–246. [Google Scholar] [CrossRef]
- Shuttleworth, T.J. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+] i oscillations. J. Biol. Chem. 1996, 271, 21720–21725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A Novel Native Store-operated Calcium Channel Encoded by Orai3 selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Dubois, C.; Abeele, F.V.; Lehen’kyi, V.y.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Kucukkaya, B.; Basoglu, H.; Erdag, D.; Akbas, F.; Susgun, S.; Yalcintepe, L. Calcium homeostasis in cisplatin resistant epithelial ovarian cancer. Gen. Physiol. Biophys. 2019, 38, 353–363. [Google Scholar] [CrossRef]
- Bergner, A.; Kellner, J.; Tufman, A.; Huber, R.M. Endoplasmic reticulum Ca 2+-homeostasis is altered in small and non-small cell lung cancer cell lines. J. Exp. Clin. Cancer Res. 2009, 28, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korošec, B.; Glavač, D.; Rott, T.; Ravnik-Glavač, M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet. Cytogenet. 2006, 171, 105–111. [Google Scholar] [CrossRef]
- Pacifico, F.; Ulianich, L.; De Micheli, S.; Treglia, S.; Leonardi, A.; Vito, P.; Formisano, S.; Consiglio, E.; Di Jeso, B. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J. Mol. Endocrinol. 2003, 30, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Varga, K.; Hollósi, A.; Pászty, K.; Hegedűs, L.; Szakács, G.; Tímár, J.; Papp, B.; Enyedi, Á.; Padányi, R. Expression of calcium pumps is differentially regulated by histone deacetylase inhibitors and estrogen receptor alpha in breast cancer cells. BMC Cancer 2018, 18, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, V.; Boivin, G.P.; Miller, M.L.; Liu, L.H.; Erwin, C.R.; Warner, B.W.; Shull, G.E. Haploinsufficiency of Atp2a2, encoding the sarco (endo) plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump, predisposes mice to squamous cell tumors via a novel mode of cancer susceptibility. Cancer Res. 2005, 65, 8655–8661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, B.; Brouland, J.-P. Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer Basic Clin. Res. 2011, 5, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Brouland, J.-P.; Gélébart, P.; Kovacs, T.; Enouf, J.; Grossmann, J.; Papp, B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am. J. Pathol. 2005, 167, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca 2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalai, G.; Krishnamurthy, R.; Hajnóczky, G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Vanoverberghe, K.; Abeele, F.V.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca 2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004, 11, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Schoneich, C.; Dremina, E.; Hewarathna, A. Bcl-2 modulates ER/SR calcium uptake by interaction with SERCA and heat shock proteins. Free Radic. Biol. Med. 2017, 108, S73. [Google Scholar] [CrossRef]
- Dremina, E.S.; Sharov, V.S.; Kumar, K.; Zaidi, A.; Michaelis, E.K.; Schöneich, C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem. J. 2004, 383, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masud, A.; Mohapatra, A.; Lakhani, S.A.; Ferrandino, A.; Hakem, R.; Flavell, R.A. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J. Biol. Chem. 2007, 282, 14132–14139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Ramirez, F.G.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharmacother. 2014, 68, 1141–1148. [Google Scholar] [CrossRef]
- Crépin, A.; Bidaux, G.; Vanden-Abeele, F.; Dewailly, E.; Goffin, V.; Prevarskaya, N.; Slomianny, C. Prolactin stimulates prostate cell proliferation by increasing endoplasmic reticulum content due to SERCA 2b over-expression. Biochem. J. 2007, 401, 49–55. [Google Scholar] [CrossRef]
- Seo, J.-A.; Kim, B.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Curcumin induces apoptosis by inhibiting sarco/endoplasmic reticulum Ca2+ ATPase activity in ovarian cancer cells. Cancer Lett. 2016, 371, 30–37. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Gou, W.-F.; Yang, X.; Wang, G.-L.; Takahashi, H.; Yu, M.; Mao, X.-Y.; Takano, Y.; Zheng, H.-C. Aberrant SERCA3 expression is closely linked to pathogenesis, invasion, metastasis, and prognosis of gastric carcinomas. Tumor Biol. 2012, 33, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.; Rao, R. Calcium-ATPases: Gene disorders and dysregulation in cancer. Biochim. Biophys. Acta 2016, 1863, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Parsonage, M.T.; Cabot, P.J.; Parat, M.-O.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int. 2013, 13, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbabian, A.; Brouland, J.P.; Gélébart, P.; Kovàcs, T.; Bobe, R.; Enouf, J.; Papp, B. Endoplasmic reticulum calcium pumps and cancer. Biofactors 2011, 37, 139–149. [Google Scholar] [CrossRef]
- Xu, M.; Seas, A.; Kiyani, M.; Ji, K.S.; Bell, H.N. A temporal examination of calcium signaling in cancer-from tumorigenesis, to immune evasion, and metastasis. Cell Biosci. 2018, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Carbone, M.; Amelio, I.; Affar, E.B.; Brugarolas, J.; Cannon-Albright, L.A.; Cantley, L.C.; Cavenee, W.K.; Chen, Z.; Croce, C.M.; D’Andrea, A.; et al. Consensus report of the 8 and 9th Weinman Symposia on Gene x Environment Interaction in carcinogenesis: Novel opportunities for precision medicine. Cell Death Differ. 2018, 25, 1885–1904. [Google Scholar] [CrossRef] [Green Version]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca 2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Tombal, B.; Weeraratna, A.T.; Denmeade, S.R.; Isaacs, J.T. Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate 2000, 43, 303–317. [Google Scholar] [CrossRef]
- Wu, Y.; Fabritius, M.; Ip, C. Chemotherapeutic sensitization by endoplasmic reticulum stress: Increasing the efficacy of taxane against prostate cancer. Cancer Biol. Ther. 2009, 8, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflügers Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, B.; Sankaranarayanan, K. Glu106 targeted inhibitors of ORAI1 as potential Ca2+ release-activated Ca2+ (CRAC) channel blockers–molecular modeling and docking studies. J. Recept. Signal Transduct. 2016, 36, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.C.; Qu, B.; Hoth, M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim. Biophys. Acta 2013, 1833, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Hoth, M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim. Biophys. Acta 2016, 1863, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Maul-Pavicic, A.; Chiang, S.C.; Rensing-Ehl, A.; Jessen, B.; Fauriat, C.; Wood, S.M.; Sjöqvist, S.; Hufnagel, M.; Schulze, I.; Bass, T.; et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc. Natl. Acad. Sci. USA 2011, 108, 3324–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-F.; Lin, P.-C.; Yeh, Y.-M.; Chen, L.-H.; Shen, M.-R. Store-operated Ca2+ entry in tumor progression: From molecular mechanisms to clinical implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebak, M.; Kinet, J.-P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, X.; Sterea, A.M.; El Hiani, Y. Lessons from the Endoplasmic Reticulum Ca2+ Transporters—A Cancer Connection. Cells 2020, 9, 1536. https://doi.org/10.3390/cells9061536
Zhai X, Sterea AM, El Hiani Y. Lessons from the Endoplasmic Reticulum Ca2+ Transporters—A Cancer Connection. Cells. 2020; 9(6):1536. https://doi.org/10.3390/cells9061536
Chicago/Turabian StyleZhai, Xingjian, Andra Mihaela Sterea, and Yassine El Hiani. 2020. "Lessons from the Endoplasmic Reticulum Ca2+ Transporters—A Cancer Connection" Cells 9, no. 6: 1536. https://doi.org/10.3390/cells9061536
APA StyleZhai, X., Sterea, A. M., & El Hiani, Y. (2020). Lessons from the Endoplasmic Reticulum Ca2+ Transporters—A Cancer Connection. Cells, 9(6), 1536. https://doi.org/10.3390/cells9061536