Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor Microenvironment
3. Tumor Neovascularization and Cancer Metastasis
3.1. Tumor Neovascularization
3.2. Hematogenous and Lymphogenous Cancer Metastases
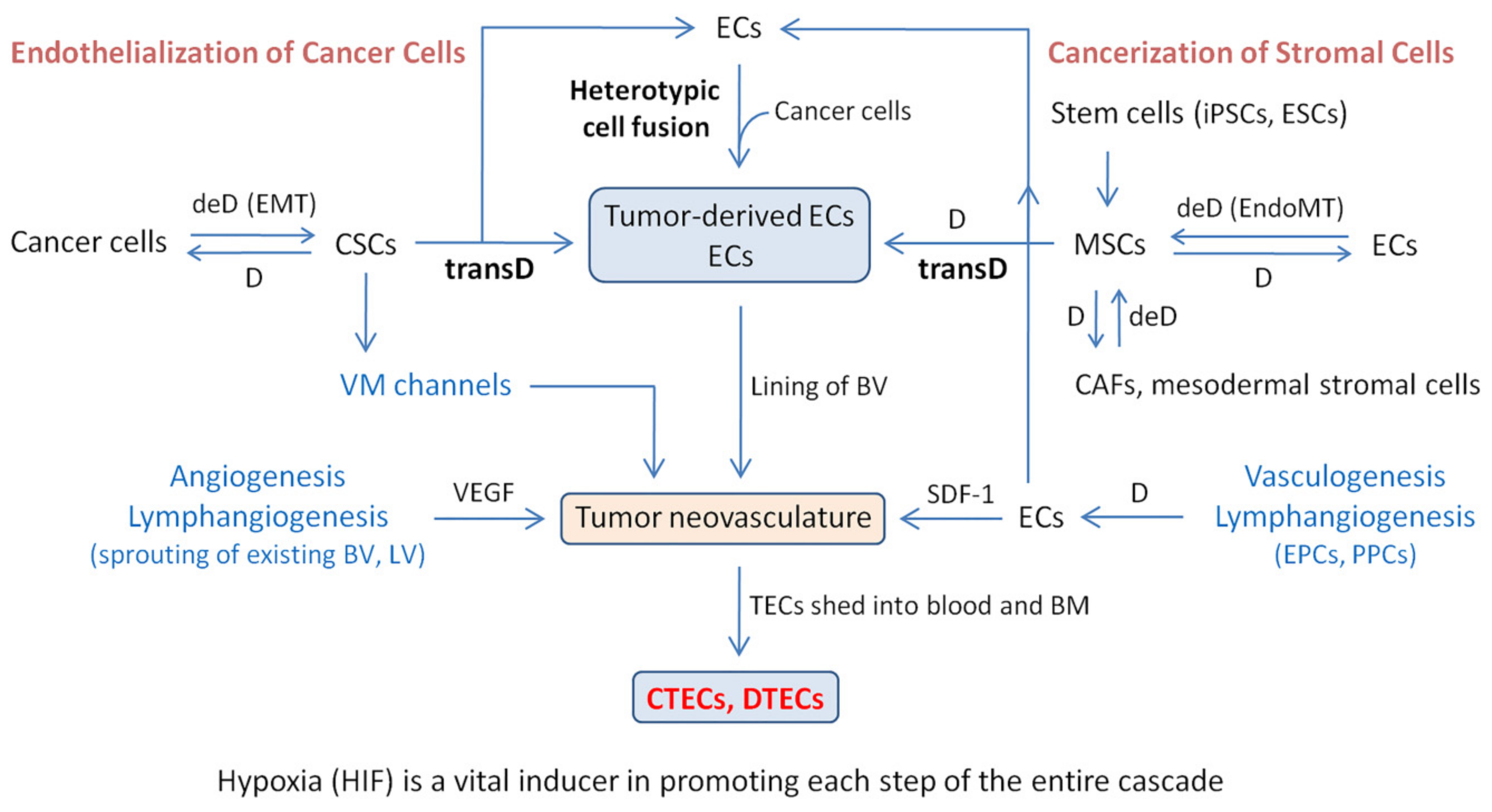
4. Mechanism of Tumor-Derived EC and CTEC Formation: Endothelialization of Malignant Cancer Cells and Cancerization of Stromal Cells
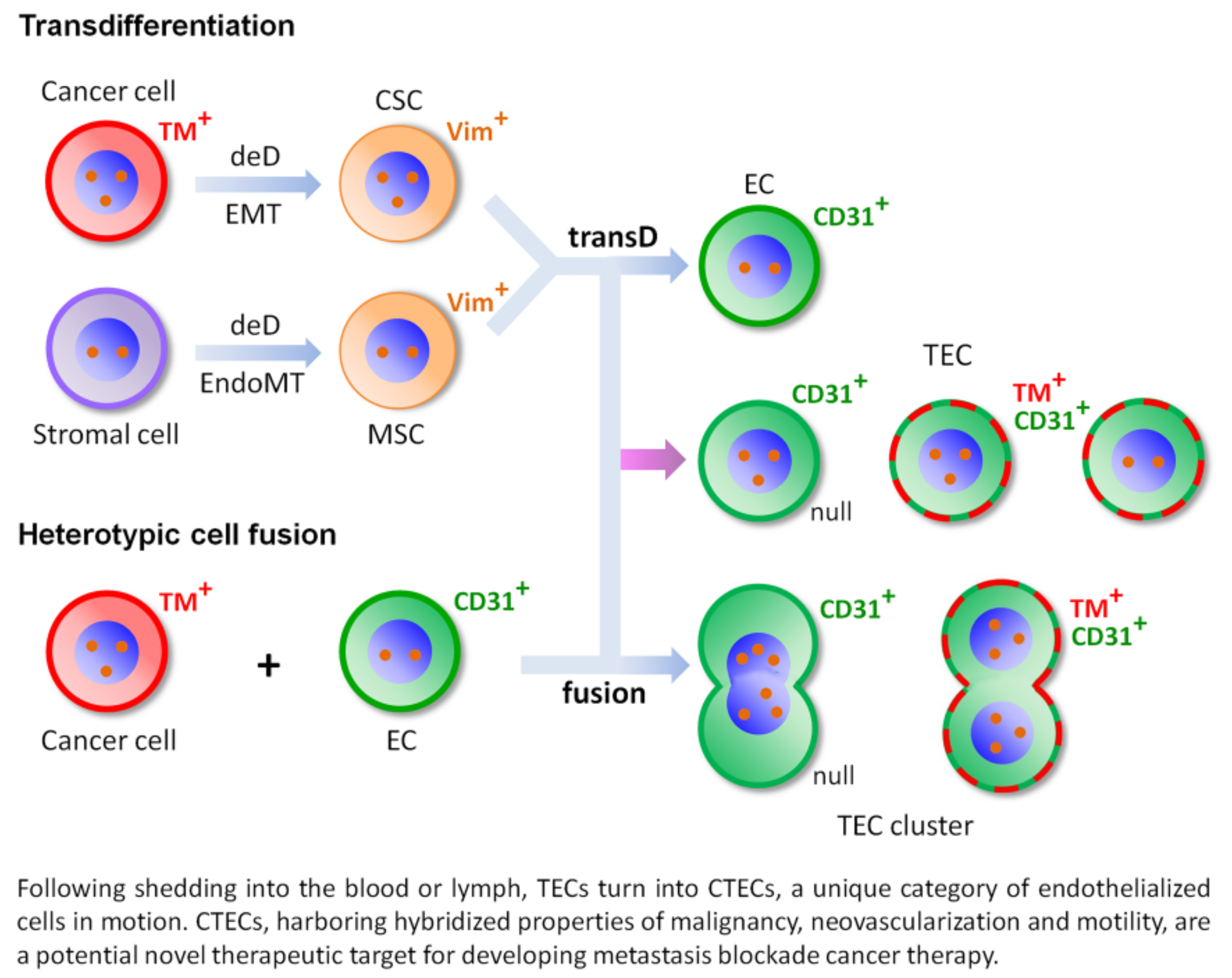
4.1. Transdifferentiation
4.2. Heterotypic Cell Fusion
4.3. Schematic Depiction of Transdifferentiation and Cell Fusion
4.4. Comprehensive Phenotypic and Karyotypic Characterization of TM+ CTECs and DTECs in Varieties of Cancer Patients
5. Hypoxia Induces Cancer Metastasis and Tumor Neovascularization
5.1. Hypoxia Induces EMT and EndoMT
5.1.1. Hypoxia-Induced EMT: An Essential Process in Cancer Metastasis and CSC Formation
5.1.2. Hypoxia-Induced EndoMT: A Critical Process in MSC and CAF Formation and Vasculature Remodeling
5.2. Hypoxia Induces Tumor Neovascularization In Vivo and Formation of Cancer Cell-Derived Vasculature In Vitro
5.3. Hypoxia-Induced Cell Fusion Participates in Tumor Neovascularization and Cancer Distant Metastasis
6. Clinical Significance and the Novel Therapeutic Target Potential of Metastatic CTECs
6.1. TECs and CTECs in Hematogenous and Lymphogenous Cancer Metastases
6.2. Co-Detection of CTC and CTEC Subtypes to Predict and Evaluate Therapeutic Efficacy of Anti-Angiogenic Regimens
6.3. CTEC: An Emerging Therapeutic Target in Motion
7. Summary
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef]
- Finger, E.C.; Giaccia, A.J. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Wu, K.J. Endothelial Transdifferentiation of Tumor Cells Triggered by the Twist1-Jagged1-KLF4 Axis: Relationship between Cancer Stemness and Angiogenesis. Stem Cells Int. 2016, 2016, 6439864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, K.; Klagsbrun, M. A new perspective on tumor endothelial cells: Unexpected chromosome and centrosome abnormalities. Cancer Res. 2005, 65, 2507–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.J.; Ravi, V.; Menter, D.G.; Sood, A.K. Endothelial cell malignancies: New insights from the laboratory and clinic. NPJ Precis. Oncol. 2017, 1, 11–13. [Google Scholar] [CrossRef]
- Lin, P.P.; Gires, O.; Wang, D.D.; Li, L.; Wang, H. Comprehensive in situ co-detection of aneuploid circulating endothelial and tumor cells. Sci. Rep. 2017, 7, 9789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, X.; Liu, Y.; Zhang, T.; Wang, Z.; Gu, M.; Li, Y.; Wang, D.D.; Li, W.; Lin, P.P. PD-L1+ aneuploid circulating tumor endothelial cells (CTECs) exhibit resistance to the checkpoint blockade immunotherapy in advanced NSCLC patients. Cancer Lett. 2020, 469, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, J.; Li, D.; Wang, Z.; Zhao, J.; Wu, X.; Sun, Q.; Lin, P.P.; Plum, P.; Damanakis, A.; et al. Tumor biology and multidisciplinary strategies of oligometastasis in gastrointestinal cancers. Semin. Cancer Biol. 2020, 60, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.P. Aneuploid CTC and CEC. Diagnostic 2018, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaks, V.; Kong, V.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Keating, A. Mesenchymal stromal cells: New directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- De Spiegelaere, W.; Casteleyn, C.; Van den Broeck, W.; Plendl, J.; Bahramsoltani, M.; Simoens, P.; Djonov, V.; Cornillie, P. Intussusceptive angiogenesis: A biologically relevant form of angiogenesis. J. Vasc. Res. 2012, 49, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad. Sci. USA 2000, 97, 14608–14613. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Brown, J.M. Vasculogenesis: A crucial player in the resistance of solid tumours to radiotherapy. Br. J. Radiol. 2014, 87, 20130686. [Google Scholar] [CrossRef] [Green Version]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, K.; Furuhata, S.; Watanabe, I.; Hayase, H.; Shimizu, A.; Ikarashi, Y.; Yoshida, T.; Terada, M.; Hashimoto, D.; Wakasugi, H. Induction of vasculogenesis in breast cancer models. Br. J. Cancer 2002, 87, 1454–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Patan, S. Vasculogenesis and angiogenesis. Cancer Treat. Res. 2004, 117, 3–32. [Google Scholar] [PubMed]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenblast, E.; Soto, M.; Gutierrez-Angel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Zhang, S.; Zhang, D.; Du, J.; Guo, H.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol. Rep. 2006, 16, 693–698. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- He, Y.; Karpanen, T.; Alitalo, K. Role of lymphangiogenic factors in tumor metastasis. Biochim. Biophys. Acta 2004, 1654, 3–12. [Google Scholar] [CrossRef]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol. 2016, 39, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Eynden, G.G.; Van der Auwera, I.; Van Laere, S.J.; Trinh, X.B.; Colpaert, C.G.; Van Dam, P.; Dirix, L.Y.; Vermeulen, P.B.; Van Marck, E.A. Comparison of molecular determinants of angiogenesis and lymphangiogenesis in lymph node metastases and in primary tumours of patients with breast cancer. J. Pathol. 2007, 213, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer 2008, 8, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Stoecklein, N.H.; Lin, P.P.; Gires, O. Circulating and disseminated tumor cells: Diagnostic tools and therapeutic targets in motion. Oncotarget 2017, 8, 1884–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Dong, Y.Y.; Wang, W.M.; Xie, X.Y.; Wang, Z.M.; Chen, R.X.; Chen, J.; Gao, D.M.; Cui, J.F.; Ren, Z.G. Vascular endothelial cells facilitated HCC invasion and metastasis through the Akt and NF-κB pathways induced by paracrine cytokines. J. Exp. Clin. Cancer Res. 2013, 32, 51. [Google Scholar] [CrossRef] [Green Version]
- Dotto, G.P. Multifocal epithelial tumors and field cancerization: Stroma as a primary determinant. J. Clin. Invest. 2014, 124, 1446–1453. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; Van den Brule, F.; Velculescu, V.; Polyak, K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef]
- Lapis, K.; Paku, S.; Liotta, L.A. Endothelialization of embolized tumor cells during metastasis formation. Clin. Exp. Metastasis 1988, 6, 73–89. [Google Scholar] [CrossRef]
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Alvero, A.B.; Fu, H.H.; Holmberg, J.; Visintin, I.; Mor, L.; Marquina, C.C.; Oidtman, J.; Silasi, D.A.; Mor, G. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells 2009, 27, 2405–2413. [Google Scholar] [CrossRef] [Green Version]
- Bussolati, B.; Bruno, S.; Grange, C.; Ferrando, U.; Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008, 22, 3696–3705. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slukvin, I.I.; Vodyanik, M. Endothelial origin of mesenchymal stem cells. Cell Cycle 2011, 10, 1370–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisan, M. Transition of mesenchymal stem/stromal cells to endothelial cells. Stem Cell Res. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, J.; Boxberger, S.; Jorgensen, B.; Feldmann, S.; Ehninger, G.; Bornhauser, M.; Werner, C. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 2004, 22, 377–384. [Google Scholar] [CrossRef]
- Gough, P.J.; Gordon, S. The role of scavenger receptors in the innate immune system. Microbes Infect. 2000, 2, 305–311. [Google Scholar] [CrossRef]
- Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte-Cancer Cell Fusion-Genesis of a Deadly Journey. Cells 2019, 8, 170. [Google Scholar] [CrossRef] [Green Version]
- Ogle, B.M.; Cascalho, M.; Platt, J.L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 2005, 6, 567–575. [Google Scholar] [CrossRef]
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef] [Green Version]
- De Baetselier, P.; Roos, E.; Brys, L.; Remels, L.; Gobert, M.; Dekegel, D.; Segal, S.; Feldman, M. Nonmetastatic tumor cells acquire metastatic properties following somatic hybridization with normal cells. Cancer Metastasis Rev. 1984, 3, 5–24. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Ogle, B.M. Cancer Cell Fusion: Mechanisms Slowly Unravel. Int. J. Mol. Sci. 2016, 17, 1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef]
- Torres, E.M.; Williams, B.R.; Amon, A. Aneuploidy: Cells losing their balance. Genetics 2008, 179, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passerini, V.; Ozeri-Galai, E.; De Pagter, M.S.; Donnelly, N.; Schmalbrock, S.; Kloosterman, W.P.; Kerem, B.; Storchova, Z. The presence of extra chromosomes leads to genomic instability. Nat. Commun. 2016, 7, 10754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duesberg, P.; Rasnick, D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil. Cytoskelet. 2000, 47, 81–107. [Google Scholar] [CrossRef]
- Wagner, M.; Hampel, B.; Bernhard, D.; Hala, M.; Zwerschke, W.; Jansen-Durr, P. Replicative senescence of human endothelial cells in vitro involves G1 arrest, polyploidization and senescence-associated apoptosis. Exp. Gerontol. 2001, 36, 1327–1347. [Google Scholar] [CrossRef]
- Mortensen, K.; Lichtenberg, J.; Thomsen, P.; Larsson, L.I. Spontaneous fusion between cancer cells and endothelial cells. Cell. Mol. Life Sci. CMLS 2004, 61, 2125–2131. [Google Scholar] [CrossRef]
- Choi, H.; Moon, A. Crosstalk between cancer cells and endothelial cells: Implications for tumor progression and intervention. Arch. Pharm. Res. 2018, 41, 711–724. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045. [Google Scholar] [CrossRef]
- Bjerregaard, B.; Holck, S.; Christensen, I.J.; Larsson, L.I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. CMLS 2006, 63, 1906–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Zhu, F.; Zhang, H.Z.; Shang, Z.J. Tumor necrosis factor-α enhanced fusions between oral squamous cell carcinoma cells and endothelial cells via VCAM-1/VLA-4 pathway. Exp. Cell Res. 2012, 318, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, R.; Von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Pesaresi, M.; Sebastian-Perez, R.; Cosma, M.P. Dedifferentiation, transdifferentiation and cell fusion: In vivo reprogramming strategies for regenerative medicine. FEBS J. 2019, 286, 1074–1093. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.P. Integrated EpCAM-independent subtraction enrichment and iFISH strategies to detect and classify disseminated and circulating tumors cells. Clin. Transl. Med. 2015, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Fischer, T.; Lavoie, C.; Huang, H.; Farquhar, M.G. Calnuc plays a role in dynamic distribution of Gαi but not Gβ subunits and modulates ACTH secretion in AtT-20 neuroendocrine secretory cells. Mol. Neurodegener. 2009, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Fischer, T.; Weiss, T.; Farquhar, M.G. Calnuc, an EF-hand Ca2+ binding protein, specifically interacts with the C-terminal α5-helix of Gαi3. Proc. Natl. Acad. Sci. USA 2000, 97, 674–679. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, X.; Liu, D.; Gong, J.; Wang, D.D.; Li, S.; Peng, Z.; Wang, X.; Lin, P.P.; Li, M.; et al. Evolutionary Expression of HER2 Conferred by Chromosome Aneuploidy on Circulating Gastric Cancer Cells Contributes to Developing Targeted and Chemotherapeutic Resistance. Clin. Cancer Res. 2018, 24, 5261–5271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, J.; Cadilha, B.L.; Markota, A.; Voigt, C.; Huang, Z.; Lin, P.P.; Wang, D.D.; Dai, J.; Kranz, G.; et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci. Adv. 2019, 5, eaav4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, Y.; Xu, J.; Zhang, A.; Wang, X.; Tang, R.; Zhang, X.; Yin, H.; Liu, M.; Wang, D.D.; et al. Quantified postsurgical small cell size CTCs and EpCAM+ circulating tumor stem cells with cytogenetic abnormalities in hepatocellular carcinoma patients determine cancer relapse. Cancer Lett. 2018, 412, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, D.D.; Yang, M.; Chen, D.; Pang, L.; Guo, S.; Cai, J.; Wery, J.P.; Li, L.; Li, H.; et al. Comprehensive characterization of chemotherapeutic efficacy on metastases in the established gastric neuroendocrine cancer patient derived xenograft model. Oncotarget 2015, 6, 15639–15651. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor β1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabieres, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell. Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maetzel, D.; Denzel, S.; Mack, B.; Canis, M.; Went, P.; Benk, M.; Kieu, C.; Papior, P.; Baeuerle, P.A.; Munz, M.; et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 2009, 11, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Kieu, C.; Mack, B.; Schmitt, B.; Zeidler, R.; Gires, O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene 2004, 23, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Gires, O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer 2007, 96, 417–423. [Google Scholar] [CrossRef]
- Chaves-Perez, A.; Mack, B.; Maetzel, D.; Kremling, H.; Eggert, C.; Harreus, U.; Gires, O. EpCAM regulates cell cycle progression via control of cyclin D1 expression. Oncogene 2013, 32, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Gires, O.; Stoecklein, N.H. Dynamic EpCAM expression on circulating and disseminating tumor cells: Causes and consequences. Cell. Mol. Life Sci. CMLS 2014, 71, 4393–4402. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Jiang, Y.; Liang, M.; Li, J.; Mao, X.; Veeramootoo, J.S.; Xia, T.; Liu, X.; Wang, S. Dynamic monitoring of CD45-/CD31+/DAPI+ circulating endothelial cells aneuploid for chromosome 8 during neoadjuvant chemotherapy in locally advanced breast cancer. Adv. Med. Oncol. 2020, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Van der Gun, B.T.; Melchers, L.J.; Ruiters, M.H.; De Leij, L.F.; McLaughlin, P.M.; Rots, M.G. EpCAM in carcinogenesis: The good, the bad or the ugly. Carcinogenesis 2010, 31, 1913–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhao, K.; Hackert, T.; Zoller, M. CD44/CD44v6 a Reliable Companion in Cancer-Initiating Cell. Maintenance and Tumor Progression. Front. Cell Dev. Biol. 2018, 6, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.; Sanchez Duffhues, G.; Ten Dijke, P.; Baker, D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 2019, 22, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Van Zonneveld, A.J.; Ten Dijke, P. Transforming growth factor β-induced endothelial-to mesenchymal transition: A switch to cardiac fibrosis? Trends. Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef] [PubMed]
- McGuire, T.F.; Sajithlal, G.B.; Lu, J.; Nicholls, R.D.; Prochownik, E.V. In vivo evolution of tumor-derived endothelial cells. PLoS ONE 2012, 7, e37138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imabayashi, H.; Mori, T.; Gojo, S.; Kiyono, T.; Sugiyama, T.; Irie, R.; Isogai, T.; Hata, J.; Toyama, Y.; Umezawa, A. Redifferentiation of dedifferentiated chondrocytes and chondrogenesis of human bone marrow stromal cells via chondrosphere formation with expression profiling by large-scale cDNA analysis. Exp. Cell Res. 2003, 288, 35–50. [Google Scholar] [CrossRef]
- Prakash, J. Cancer-Associated Fibroblasts: Perspectives in Cancer Therapy. Trends Cancer 2016, 2, 277–279. [Google Scholar] [CrossRef]
- Gasparics, A.; Rosivall, L.; Krizbai, I.A.; Sebe, A. When the endothelium scores an own goal: Endothelial cells actively augment metastatic extravasation through endothelial-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1055–H1063. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Kim, A.R.; Nam, J.K.; Kim, J.M.; Kim, J.Y.; Seo, H.R.; Lee, H.J.; Cho, J.; Lee, Y.J. Tumour vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6+ cancer cell and macrophage polarization. Nat. Commun. 2018, 9, 5108. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene 2013, 32, 4057–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.L.; Sainson, R.C.; Oon, C.E.; Turley, H.; Leek, R.; Sheldon, H.; Bridges, E.; Shi, W.; Snell, C.; Bowden, E.T.; et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res. 2011, 71, 6073–6083. [Google Scholar] [CrossRef] [Green Version]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Bottinger, E.P. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Greenfield, J.P.; Cobb, W.S.; Lyden, D. Resisting arrest: A switch from angiogenesis to vasculogenesis in recurrent malignant gliomas. J. Clin. Invest. 2010, 120, 663–667. [Google Scholar] [CrossRef]
- Sun, T.; Zhao, N.; Zhao, X.L.; Gu, Q.; Zhang, S.W.; Che, N.; Wang, X.H.; Du, J.; Liu, Y.X.; Sun, B.C. Expression and functional significance of Twist1 in hepatocellular carcinoma: Its role in vasculogenic mimicry. Hepatology 2010, 51, 545–556. [Google Scholar] [CrossRef]
- Liu, Z.; Qi, L.; Li, Y.; Zhao, X.; Sun, B. VEGFR2 regulates endothelial differentiation of colon cancer cells. BMC Cancer 2017, 17, 593. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, W.; Wang, S.; Yu, F.; Feng, J.; Wang, X.; Zhang, L.; Lin, J. Transdifferentiation of human MNNG/HOS osteosarcoma cells into vascular endothelial cells in vitro and in vivo. Oncol. Rep. 2017, 38, 3153–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.J.; Gao, J.; Ma, X.B.; Yan, K.; Liu, X.X.; Kang, H.F.; Ji, Z.Z.; Guan, H.T.; Wang, X.J. Up-regulation of hypoxia inducible factor-1α by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells. J. Exp. Clin. Cancer Res. 2012, 31, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Huang, G.; Li, X.; Zhang, Y.; Jiang, Y.; Shen, J.; Liu, J.; Wang, Q.; Zhu, J.; Feng, X.; et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor-1α in hepatocellular carcinoma. BMC Cancer 2013, 13, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Schmid, T.; Brune, B. Tumor necrosis factor-α causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. Mol. Biol. Cell 2003, 14, 2216–2225. [Google Scholar] [CrossRef]
- Zhan, J.; Li, Y.; Yu, J.; Zhao, Y.; Cao, W.; Ma, J.; Sun, X.; Sun, L.; Qian, H.; Zhu, W.; et al. Culture medium of bone marrow-derived human mesenchymal stem cells effects lymphatic endothelial cells and tumor lymph vessel formation. Oncol. Lett. 2015, 9, 1221–1226. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Jubb, A.M.; Harris, A.L. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol. 2010, 11, 1172–1183. [Google Scholar] [CrossRef]
- Bidard, F.C.; Mathiot, C.; Degeorges, A.; Etienne-Grimaldi, M.C.; Delva, R.; Pivot, X.; Veyret, C.; Bergougnoux, L.; De Cremoux, P.; Milano, G.; et al. Clinical value of circulating endothelial cells and circulating tumor cells in metastatic breast cancer patients treated first line with bevacizumab and chemotherapy. Ann. Oncol. 2010, 21, 1765–1771. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. CMLS 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Yeldag, G.; Rice, A.; Del Rio Hernandez, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.Q.; Sun, H.C.; Zhang, W.; Zhu, X.D.; Zhuang, P.Y.; Zhang, J.B.; Wang, L.; Wu, W.Z.; Qin, L.X.; Tang, Z.Y. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Sandhu, S.K.; Workman, P.; De Bono, J.S. Envisioning the future of early anticancer drug development. Nat. Rev. Cancer 2010, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Zhang, X.T.; Ge, S.; Gao, J.; Gong, J.F.; Lu, M.; Zhang, Q.Y.; Cao, Y.S.; Wang, D.D.; Lin, P.P.; et al. Clinical significance of phenotyping and karyotyping of circulating tumor cells in patients with advanced gastric cancer. Oncotarget 2014, 5, 6594–6602. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Zhang, L.; Tong, L.; Gao, Y.; Hu, F.; Lin, P.P.; Li, B.; Zhang, T. Vimentin expression in circulating tumor cells (CTCs) associated with liver metastases predicts poor progression-free survival in patients with advanced lung cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2911–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchado, E.; Malumbres, M. Targeting aneuploidy for cancer therapy. Cell 2011, 144, 465–466. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, P.P. Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis. Cells 2020, 9, 1539. https://doi.org/10.3390/cells9061539
Lin PP. Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis. Cells. 2020; 9(6):1539. https://doi.org/10.3390/cells9061539
Chicago/Turabian StyleLin, Peter Ping. 2020. "Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis" Cells 9, no. 6: 1539. https://doi.org/10.3390/cells9061539
APA StyleLin, P. P. (2020). Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis. Cells, 9(6), 1539. https://doi.org/10.3390/cells9061539