A Review on Notch Signaling and Colorectal Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
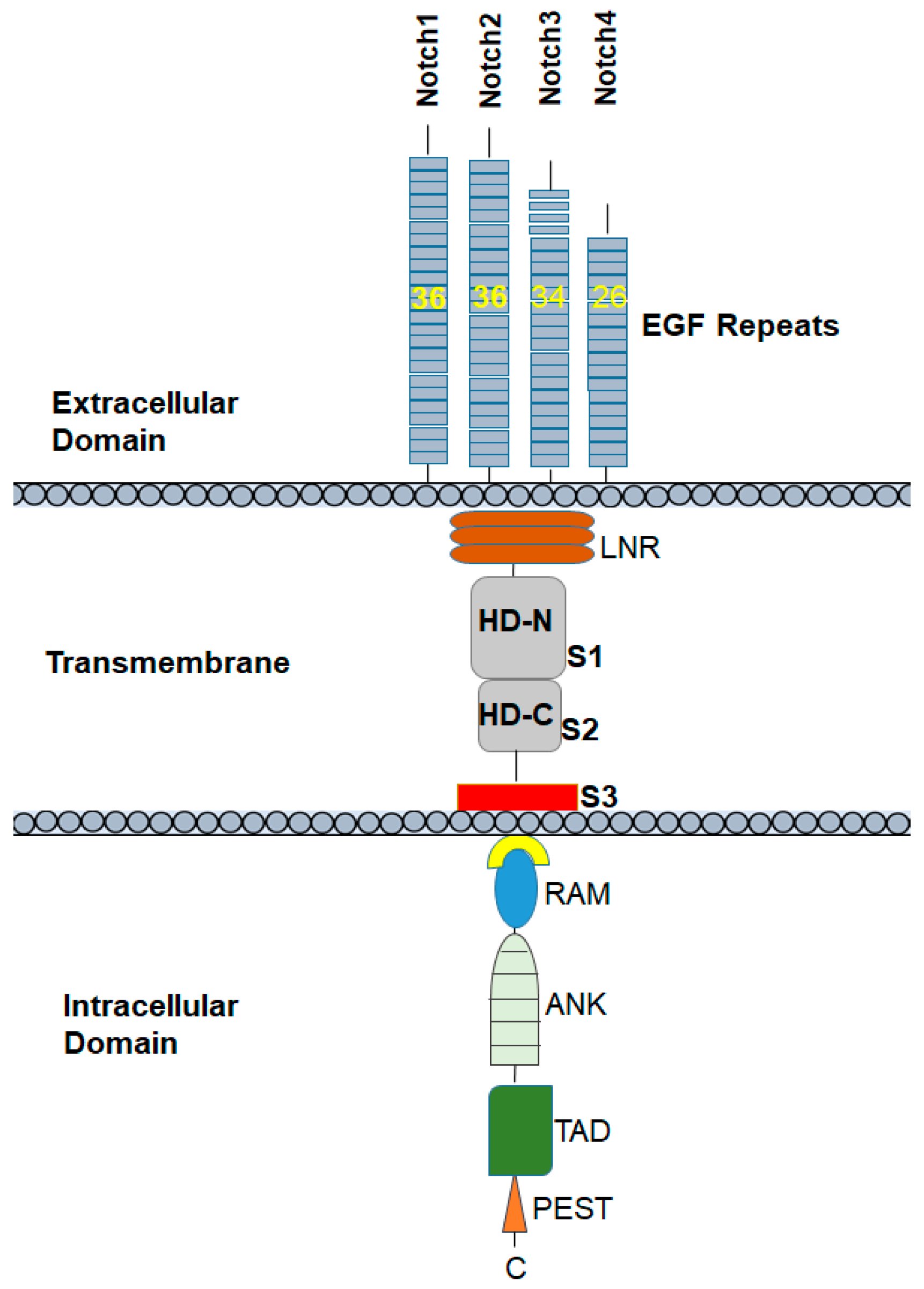
2. Notch Receptors: Structure and Function
3. Notch Receptors Have Structural and Functional Differences
4. The Role of Notch1 in the Proliferation and EMT
5. Notch1 Signaling in Colorectal Cancer
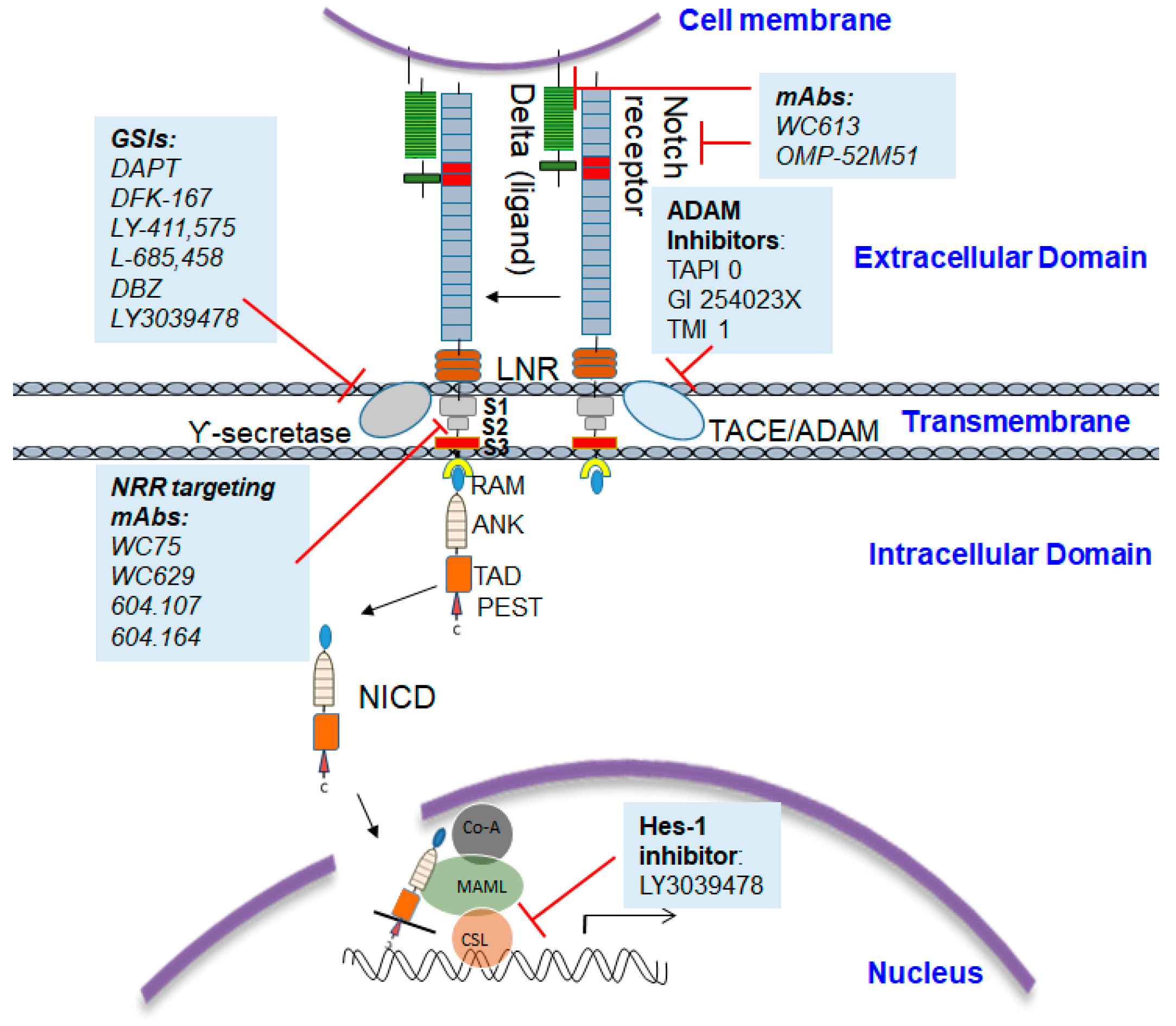
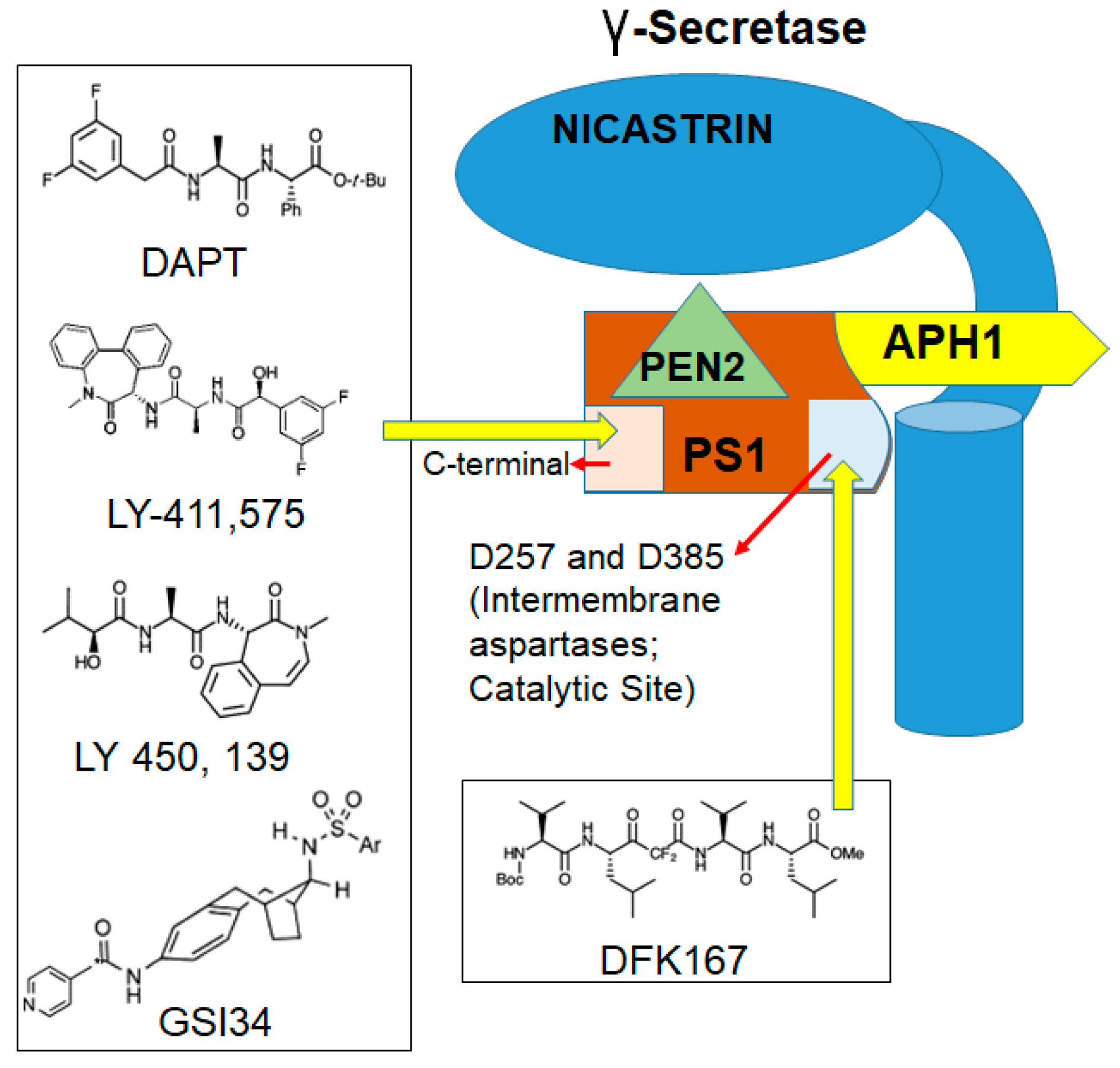
6. Small Molecule Inhibitors of Notch1 Signaling
7. Natural Compounds Target Notch1 Activation
8. Targeting Downstream Notch1 Signaling Activators
9. Limitations of GSIs and mAbs in Notch1 Treatment
10. Is NRR-Notch1 an Ideal Target for Therapy?
11. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chien, C.-R.C.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, P.J.; Sundaresan, S.R. Surgical Management of Colorectal Lung Metastasis. Clin. Colon Rectal Surg. 2009, 22, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J. Claudin-1 enhances tumor proliferation and metastasis by regulating cell anoikis in gastric cancer. Oncotarget 2015, 6, 1652–1665. [Google Scholar] [CrossRef] [Green Version]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch Activation Results in Phenotypic and Functional Changes Consistent With Endothelial-to-Mesenchymal Transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Kemper, K.; Grandela, C.; Medema, J.P. Molecular identification and targeting of colorectal cancer stem cells. Oncotarget 2010, 1, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Rosenberg, D.W. Role of Notch signaling in colon homeostasis and carcinogenesis. Cancer Sci. 2011, 102, 1938–1942. [Google Scholar] [CrossRef]
- Geissler, K.; Zach, O. Pathways involved in Drosophila and human cancer development: The Notch, Hedgehog, Wingless, Runt, and Trithorax pathway. Ann. Hematol. 2012, 91, 645–669. [Google Scholar] [CrossRef]
- Meng, R.D.; Shelton, C.C.; Li, Y.-M.; Qin, L.-X.; Notterman, D.; Paty, P.B.; Schwartz, G.K. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009, 69, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, B.; Pal, D.; Kolluru, V.; Tyagi, A.; Baby, B.; Dahiya, N.R.; Youssef, K.; Alatassi, H.; Ankem, M.K.; Sharma, A.K.; et al. The chemopreventive effect of withaferin A on spontaneous and inflammation-associated colon carcinogenesis models. Carcinogenesis 2018, 39, 1537–1547. [Google Scholar] [CrossRef]
- Pal, D.; Tyagi, A.; Chandrasekaran, B.; Alattasi, H.; Ankem, M.K.; Sharma, A.K.; Damodaran, C. Suppression of Notch1 and AKT mediated epithelial to mesenchymal transition by Verrucarin J in metastatic colon cancer. Cell Death Dis. 2018, 9, 798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortini, M.E.; Rebay, I.; Caron, L.A.; Artavanis-Tsakonas, S. An activated Notch receptor blocks cell-fate commitment in the developing Drosophila eye. Nature 1993, 365, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Pourquie, O. Segmentation of the paraxial mesoderm and vertebrate somitogenesis. Curr. Top. Dev. Biol. 2000, 47, 81–105. [Google Scholar] [PubMed]
- Gridley, T. Notch signaling during vascular development. Proc. Natl. Acad. Sci. USA 2001, 98, 5377–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, Á.; Bertran, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo-Ponce, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genome Res. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Smith, S.M.; Hu, X. Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell 2016, 7, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Fre, S.; Huyghe, M.; Mourikis, P.; Robine, S.; Louvard, D.; Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968. [Google Scholar] [CrossRef]
- Van Es, J.H.; Clevers, H. Notch and Wnt inhibitors as potential new drugs for intestinal neoplastic disease. Trends Mol. Med. 2005, 11, 496–502. [Google Scholar] [CrossRef]
- Okamoto, R.; Tsuchiya, K.; Nemoto, Y.; Akiyama, J.; Nakamura, T.; Kanai, T.; Watanabe, M. Requirement of Notch activation during regeneration of the intestinal epithelia. Am. J. Physiol. Liver Physiol. 2009, 296, G23–G35. [Google Scholar] [CrossRef] [Green Version]
- Radtke, F.; Clevers, H.; Riccio, O. From gut homeostasis to cancer. Curr. Mol. Med. 2006, 6, 275–289. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, L.; Moens, C.B.; Appel, B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development 2013, 141, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyttendaele, H.; Marazzi, G.; Wu, G.; Yan, Q.; Sassoon, D.; Kitajewski, J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development 1996, 122, 2251–2259. [Google Scholar] [PubMed]
- Liu, Z.; Brunskill, E.; Varnum-Finney, B.; Zhang, C.; Zhang, A.; Jay, P.Y.; Bernstein, I.; Morimoto, M.; Kopan, R. The intracellular domains of Notch1 and Notch2 are functionally equivalent during development and carcinogenesis. Development 2015, 142, 2452–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, W.R.; Roy, M.; Ulu, D.V.; Garfinkel, M.B.; Mansour, M.R.; Aster, J.C.; Blacklow, S.C. Structure of the Notch1-negative regulatory region: Implications for normal activation and pathogenic signaling in T-ALL. Blood 2009, 113, 4381–4390. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.K.K.; Wang, X.J.; Cheng, A.; Luo, M.X.; Ng, S.S.; To, K.F.; Chan, F.K.L.; Cho, C.H.; Sung, J.J.Y.; Yu, J. Dysregulation and crosstalk of cellular signaling pathways in colon carcinogenesis. Crit. Rev. Oncol. 2013, 86, 251–277. [Google Scholar] [CrossRef]
- Huang, T.; Zhou, Y.; Cheng, A.; Yu, J.; To, K.-F.; Kang, W. NOTCH receptors in gastric and other gastrointestinal cancers: Oncogenes or tumor suppressors? Mol. Cancer 2016, 15, 80. [Google Scholar] [CrossRef] [Green Version]
- Revandkar, A.; Perciato, M.L.; Toso, A.; Alajati, A.; Chen, J.; Gerber, H.; Dimitrov, M.; Rinaldi, A.; Delaleu, N.; Pasquini, E.; et al. Inhibition of Notch pathway arrests PTEN-deficient advanced prostate cancer by triggering p27-driven cellular senescence. Nat. Commun. 2016, 7, 13719. [Google Scholar] [CrossRef]
- Hristova, N.R.; Tagscherer, K.E.; Fassl, A.; Kopitz, J.; Roth, W. Notch1-dependent regulation of p27 determines cell fate in colorectal cancer. Int. J. Oncol. 2013, 43, 1967–1975. [Google Scholar] [CrossRef]
- Bol, G.; Raman, V.; Van Der Groep, P.; Vermeulen, J.F.; Patel, A.H.; Van Der Wall, E.; Van Diest, P.J. Expression of the RNA Helicase DDX3 and the Hypoxia Response in Breast Cancer. PLoS ONE 2013, 8, e63548. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Osborne, B.A. Arbiter of differentiation and death: Notch signaling meets apoptosis. J. Cell. Physiol. 1999, 181, 393–409. [Google Scholar] [CrossRef]
- Gan, R.-H.; Wei, H.; Xie, J.; Zheng, D.-P.; Luo, E.-L.; Huang, X.-Y.; Xie, J.; Zhao, Y.; Ding, L.-C.; Su, B.-H.; et al. Notch1 regulates tongue cancer cells proliferation, apoptosis and invasion. Cell Cycle 2018, 17, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäättelä, M. Escaping Cell Death: Survival Proteins in Cancer. Exp. Cell Res. 1999, 248, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Sriuranpong, V.; Borges, M.W.; Ravi, R.K.; Arnold, D.R.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001, 61, 3200–3205. [Google Scholar] [PubMed]
- Gestblom, C.; Grynfeld, A.; Ora, I.; Ortoft, E.; Larsson, C.; Axelson, H.; Sandstedt, B.; Cserjesi, P.; Olson, E.N.; Påhlman, S. The basic helix-loop-helix transcription factor dHAND, a marker gene for the developing human sympathetic nervous system, is expressed in both high- and low-stage neuroblastomas. Lab. Investig. 1999, 79, 67–79. [Google Scholar] [PubMed]
- Talora, C. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev. 2002, 16, 2252–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Kunnimalaiyaan, M.; Drenzek, J.; Seiler, N. Notch 1 signaling is active in ovarian cancer. Gynecol. Oncol. 2010, 117, 130–133. [Google Scholar] [CrossRef]
- Shou, J.; Ross, S.; Koeppen, H.; De Sauvage, F.J.; Gao, W.-Q. Dynamics of notch expression during murine prostate development and tumorigenesis. Cancer Res. 2001, 61, 7291–7297. [Google Scholar]
- Allenspach, E.J.; Maillard, I.; Aster, J.C.; Pear, W.S. Notch Signaling in Cancer. Cancer Biol. Ther. 2002, 1, 466–476. [Google Scholar] [CrossRef]
- Radtke, F.; Raj, K. The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat. Rev. Cancer 2003, 3, 756–767. [Google Scholar] [CrossRef]
- Maillard, I.; Pear, W.S. Notch and cancer: Best to avoid the ups and downs. Cancer Cell 2003, 3, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Weng, A.P. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Gradilone, A.; Naso, G.; Vincenzi, B.; Petracca, A.; Nicolazzo, C.; Palazzo, A.; Saltarelli, R.; Spremberg, F.; Cortesi, E.; et al. Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res. Treat. 2011, 130, 449–455. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [Green Version]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Laé, M.; Janssen, K.-P.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazio, C.; Piazzi, G.; Vitaglione, P.; Fogliano, V.; Munarini, A.; Prossomariti, A.; Milazzo, M.; D’Angelo, L.; Napolitano, M.; Chieco, P.; et al. Inflammation increases NOTCH1 activity via MMP9 and is counteracted by Eicosapentaenoic Acid-free fatty acid in colon cancer cells. Sci. Rep. 2016, 6, 20670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, À.; Fernández-Majada, V.; Grilli, A.; Lopez-Bigas, N.; Bellora, N.; Albà, M.M.; Torres, F.; et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcaroli, J.J.; Tai, W.; McWilliams, R.; Bagby, S.; Blatchford, P.J.; Varella-Garcia, M.; Purkey, A.; Quackenbush, K.S.; Song, E.-K.; Pitts, T.M.; et al. A NOTCH1 gene copy number gain is a prognostic indicator of worse survival and a predictive biomarker to a Notch1 targeting antibody in colorectal cancer. Int. J. Cancer 2015, 138, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reedijk, M.; Odorcic, S.; Zhang, H.; Chetty, R.; Tennert, C.; Dickson, B.C.; Lockwood, G.; Gallinger, S.; Egan, S.E. Activation of Notch signaling in human colon adenocarcinoma. Int. J. Oncol. 2008, 33, 1223–1229. [Google Scholar] [PubMed] [Green Version]
- Jackstadt, R.; Van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336.e7. [Google Scholar] [CrossRef] [Green Version]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J. Cell. Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef]
- Wolfe, M.S. Gamma-secretase inhibition and modulation for Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 158–164. [Google Scholar] [CrossRef]
- Siemers, E.R. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology 2006, 66, 602–604. [Google Scholar] [CrossRef]
- Hyde, L.A. Studies to investigate the in vivo therapeutic window of the gamma-secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-di hydro-5H-dibenzo[b,d]azepin-7-yl]-L-alaninamide (LY411,575) in the CRND8 mouse. J. Pharmacol. Exp. Ther. 2006, 319, 1133–1143. [Google Scholar] [CrossRef]
- Akiyoshi, T. Gamma-secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology 2008, 134, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, T.; Feller, S.M. Gamma-secretase inhibition combined with platinum compounds enhances cell death in a large subset of colorectal cancer cells. Cell Commun. Signal. 2008, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massard, C.; Azaro, A.; Soria, J.-C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.; et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinet, L.; Rodilla, V.; Liu, Z.; Chen, S.; Koch, U.; Espinosa, L.; Kaestner, K.H.; Kopan, R.; Lewis, J.; Radtke, F. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 2011, 140, 1230–1240.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z. Inhibition of nuclear factor kappab activity by genistein is mediated via Notch-1 signaling pathway in pancreatic cancer cells. Int. J. Cancer 2006, 118, 1930–1936. [Google Scholar] [CrossRef]
- Kallifatidis, G.; Labsch, S.; Rausch, V.; Mattern, J.; Gladkich, J.; Moldenhauer, G.; Büchler, M.W.; Salnikov, A.V.; Herr, I. Sulforaphane Increases Drug-mediated Cytotoxicity Toward Cancer Stem-like Cells of Pancreas and Prostate. Mol. Ther. 2010, 19, 188–195. [Google Scholar] [CrossRef]
- Kawahara, T.; Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Cyclopamine and quercetin suppress the growth of leukemia and lymphoma cells. Anticancer. Res. 2009, 29, 4629–4632. [Google Scholar]
- Wang, Z.; Zhang, Y.; Banerjee, S.; Li, Y.; Sarkar, F.H. Retracted: Notch-1 down-regulation by curcumin is associated with the inhibition of cell growth and the induction of apoptosis in pancreatic cancer cells. Cancer 2006, 106, 2503–2513. [Google Scholar] [CrossRef]
- Cecchinato, V.; Chiaramonte, R.; Nizzardo, M.; Cristofaro, B.; Basile, A.; Sherbet, G.V.; Comi, P. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem. Pharmacol. 2007, 74, 1568–1574. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, B.-W. Withaferin-A Inhibits Colon Cancer Cell Growth by Blocking STAT3 Transcriptional Activity. J. Cancer Prev. 2015, 20, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.; Kolluru, V.; Chandrasekaran, B.; Baby, B.V.; Aman, M.; Suman, S.; Sirimulla, S.; Sanders, M.A.; Alatassi, H.; Ankem, M.K.; et al. Targeting aberrant expression of Notch-1 in ALDH+cancer stem cells in breast cancer. Mol. Carcinog. 2017, 56, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ke, J.; He, Z.; Chen, Z.; Huang, Q.; Ai, W.; Wang, G.; Wei, Y.; Zou, X.; Zhang, S.; et al. HES1 Promotes Colorectal Cancer Cell Resistance To 5-Fu by Inducing Of EMT and ABC Transporter Proteins. J. Cancer 2017, 8, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.; Tsao, P.-N.; Lin, H.; Tung, C.; Change, M.; Chang, Y.-C.; Wong, J.; Wei, S. Hes1 Increases the Invasion Ability of Colorectal Cancer Cells via the STAT3-MMP14 Pathway. PLoS ONE 2015, 10, e0144322. [Google Scholar] [CrossRef] [PubMed]
- Rohena-Rivera, K.; Bhowmick, N.A. Notch inhibitor screening reveals an unexpected HES1 heterodimer. J. Biol. Chem. 2018, 293, 8295–8296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Li, G.; You, Y.; Wan, H.; Wu, Q.; Wang, C.; Lu, N. Antitumor activity of Notch-1 inhibition in human colorectal carcinoma cells. Oncol. Rep. 2017, 39, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Zhang, Y.-C.; Peng, X.; Long, Z.; Ming, Y.; He, L.-Y. siRNA-mediated silencing of Notch-1 enhances docetaxel induced mitotic arrest and apoptosis in prostate cancer cells. Asian Pac. J. Cancer Prev. 2012, 13, 2485–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Jiang, H.; Chen, L.; Liu, J.; Hu, X.; Zhang, H. Inhibition of Notch1/Hes1 signaling pathway improves radiosensitivity of colorectal cancer cells. Eur. J. Pharmacol. 2018, 818, 364–370. [Google Scholar] [CrossRef]
- Zhou, W.; Tan, W.; Huang, X.; Yu, H.G. Doxorubicin combined with Notch1-targeting siRNA for the treatment of gastric cancer. Oncol. Lett. 2018, 16, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; De Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Li, Y.; Burns, J.A.; Cheney, C.A.; Zhang, N.; Vitelli, S.; Wang, F.; Bett, A.; Chastain, M.; Audoly, L.P.; Zhang, Z.-Q. Distinct expression profiles of Notch-1 protein in human solid tumors: Implications for development of targeted therapeutic monoclonal antibodies. Biol. Targets Ther. 2010, 4, 163–171. [Google Scholar]
- Grosveld, G.C. Gamma-secretase inhibitors: Notch so bad. Nat. Med. 2009, 15, 20–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Wong, B.C. Role of Notch signaling in colorectal cancer. Carcinogenesis 2009, 30, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.; et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [Green Version]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.; Ciaccio, P.J. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Eckhardt, G.; Patnaik, A.; Lorusso, P.; Faoro, L.; Heymach, J.; Kapoun, A.; Xu, L.; Munster, P. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann. Oncol. 2018, 29, 1561–1568. [Google Scholar] [CrossRef]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.M.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B.I. A Phase I Dose Escalation and Expansion Study of the Anticancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.; Falk, A.; Dyson, M.; Melidoni, A.; Parthiban, K.; Young, J.L.; Roake, W.; McCafferty, J. Generation of anti-Notch antibodies and their application in blocking Notch signalling in neural stem cells. Methods 2012, 58, 69–78. [Google Scholar] [CrossRef]
- Li, K.; Li, Y.; Wu, W.; Gordon, W.R.; Chang, D.W.; Lu, M.; Scoggin, S.; Fu, T.; Vien, L.; Histen, G.; et al. Modulation of Notch Signaling by Antibodies Specific for the Extracellular Negative Regulatory Region of NOTCH3. J. Biol. Chem. 2008, 283, 8046–8054. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Gadkari, R.A.; Ramakanth, S.V.; Padmanabhan, K.; Madhumathi, D.S.; Devi, L.; Appaji, L.; Aster, J.C.; Rangarajan, A.; Dighe, R.R. A novel Monoclonal Antibody against Notch1 Targets Leukemia-associated Mutant Notch1 and Depletes Therapy Resistant Cancer Stem Cells in Solid Tumors. Sci. Rep. 2015, 5, 11012. [Google Scholar] [CrossRef] [Green Version]
- Aste-Amézaga, M.; Zhang, N.; Lineberger, J.E.; Arnold, B.A.; Toner, T.J.; Gu, M.; Huang, L.; Vitelli, S.; Vo, K.T.; Haytko, P.; et al. Characterization of Notch1 Antibodies That Inhibit Signaling of Both Normal and Mutated Notch1 Receptors. PLoS ONE 2010, 5, e9094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, T.; Yan, J.; Wang, X.; Sheng, J. Secretory expression of negative regulatory region of human Notch1 in Escherichia coli and preparation of a functional polyclonal antibody. Biotechnol. Appl. Biochem. 2018, 65, 554–559. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. https://doi.org/10.3390/cells9061549
Tyagi A, Sharma AK, Damodaran C. A Review on Notch Signaling and Colorectal Cancer. Cells. 2020; 9(6):1549. https://doi.org/10.3390/cells9061549
Chicago/Turabian StyleTyagi, Ashish, Arun K. Sharma, and Chendil Damodaran. 2020. "A Review on Notch Signaling and Colorectal Cancer" Cells 9, no. 6: 1549. https://doi.org/10.3390/cells9061549
APA StyleTyagi, A., Sharma, A. K., & Damodaran, C. (2020). A Review on Notch Signaling and Colorectal Cancer. Cells, 9(6), 1549. https://doi.org/10.3390/cells9061549