Amyloid Proteins and Peripheral Neuropathy

 and
and
Abstract
:1. Introduction
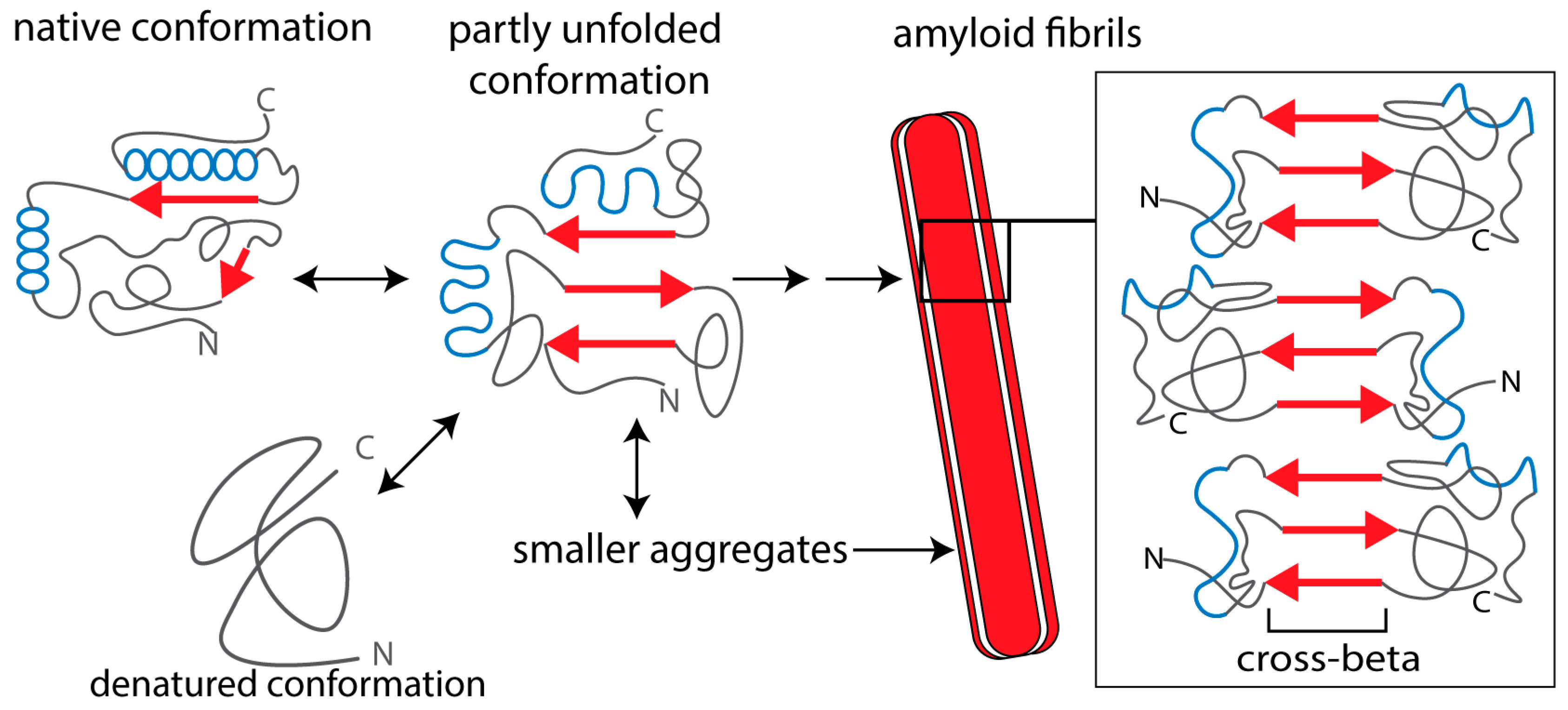
2. Causes of Amyloidogenesis
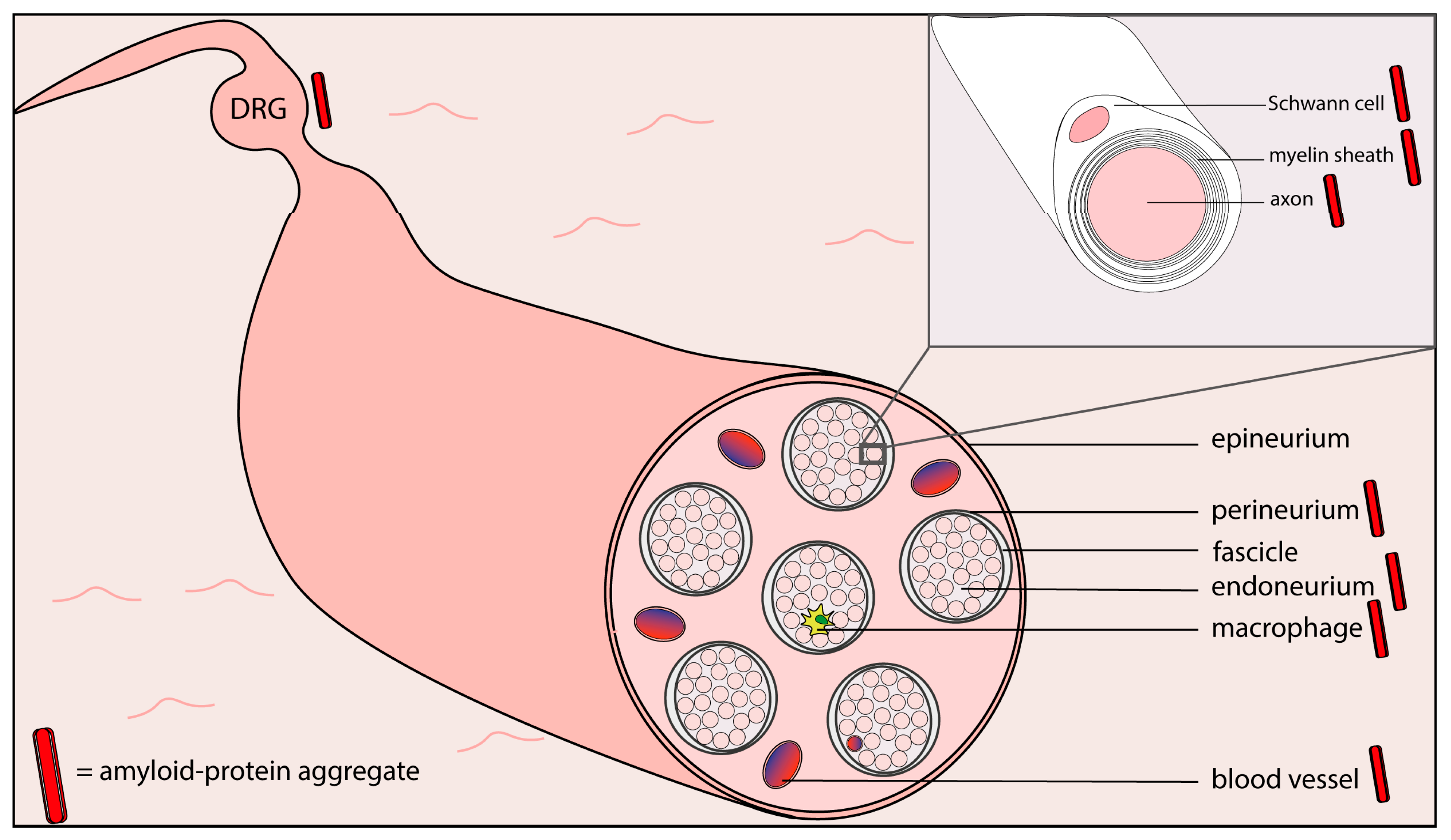
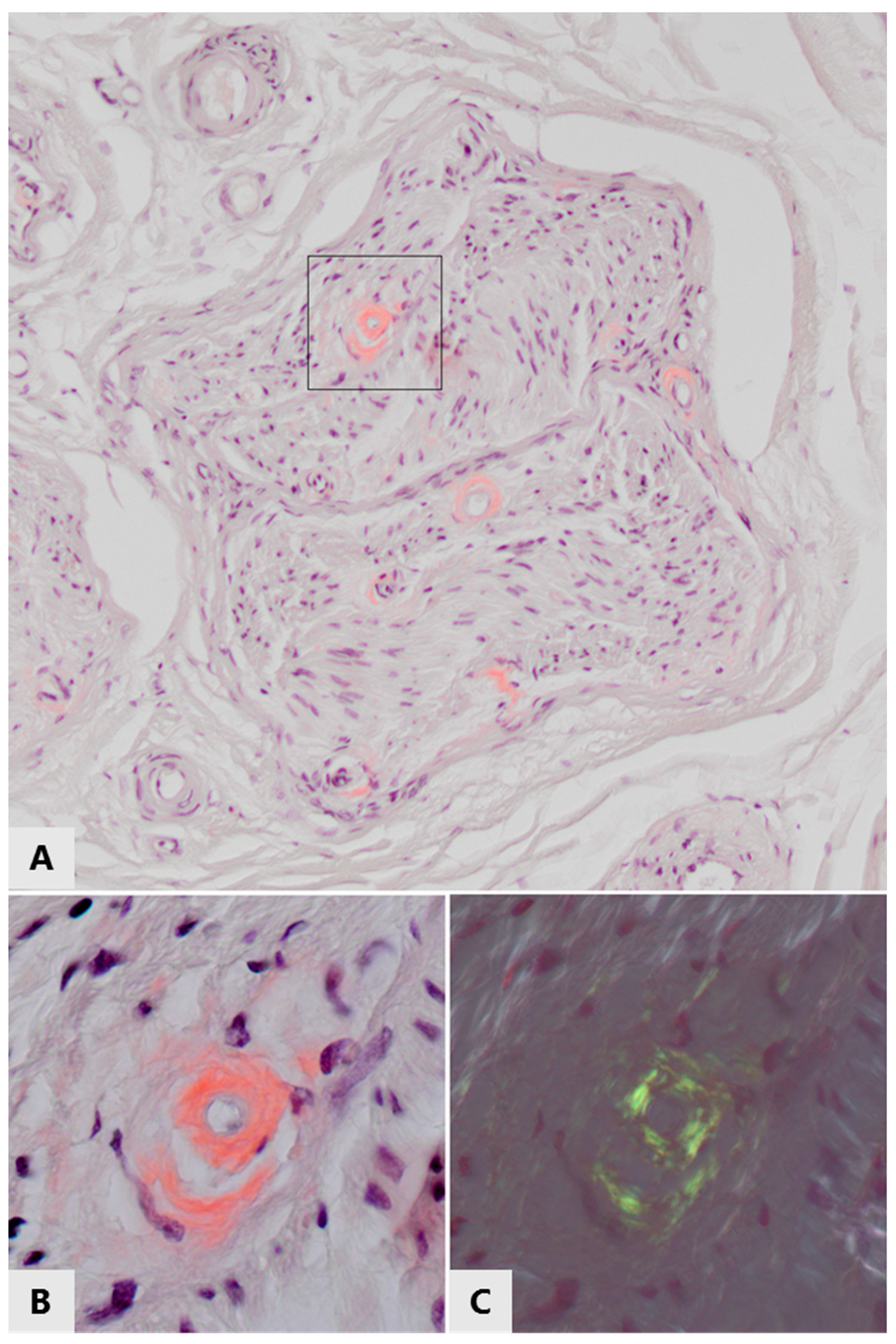
3. Peripheral Amyloid Neuropathies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Amyloid Protein | Acquired/ Hereditary | Local/ Systemic | Peripheral Nervous System Involvement | Prevalence/Incidence Disease | Prevalence Disease w/w * | Prevalence/Incidence PN (% of Patients) |
|---|---|---|---|---|---|---|---|
| Familial amyloid polyneuropathy | Transthyretin (hATTR) | Hereditary | Systemic | Polyneuropathy Autonomic disturbances Carpal tunnel syndrome [41] | 10,186 persons w/w (range: 5526–38,468)/UN [42] | 0.00013% | UN (it develops in the majority of patients/UN [42,43] |
| Apolipoprotein A-I (AApoAI) | Hereditary | Systemic | Polyneuropathy [44] | UN/UN | UN | UN/UN | |
| Gelsolin (HGA) | Hereditary | Systemic | Cranial neuropathy Polyneuropathy [28] | 400 to 1000 gene carriers in Finland/UN [45] | 0.01% | UN/UN | |
| Immunoglobulin light-chain amyloidosis | Ig light-chain | Acquired | Systemic | Polyneuropathy Autonomic disturbances Carpal tunnel syndrome [46,47] | 40.5 cases per million in the US/9.7 to 14.0 cases per million per year in US [48] | 0.004% | 15–20%/UN [49] |
| Dialysis-related amyloidosis | β2-microglobulin | Acquired | Systemic | Carpal tunnel syndrome Polyneuropathy [50,51] | UN/UN (incidence > 95% in. patients > 15 years dialysis in US) [51] | UN | UN/UN |
| Senile systemic amyloidosis | Transthyretin | Acquired | Systemic | Polyneuropathy Autonomic disturbances carpal tunnel syndrome [52] | 63/256 of the study population in Finland (25% > 80 years old)/UN [53] | 0.45% | UN/UN |
| Type 2 diabetes mellitus | IAPP | Acquired | Local/systemic | Polyneuropathy [54] | 463 million persons (aged 20–79 years) w/w (including T1DM&T2DM)/UN [55] | 5.4% | 31.5–45% [56,57] |
| Rheumatoid arthritis | SAA | Acquired | Systemic | Polyneuropathy [58] | 19,965,115 persons w/w/1,204,599 new cases w/w [59] | 0.26% | 39.19–75.28% [58] |
| Inflammatory bowel disease | SAA | Acquired | Systemic | Polyneuropathy [60,61] | 68 million persons w/w/70,000 new cases per year in USA [62,63] | 0.09% | UN/0.07% after 10 years of IBD [60,64] |
| Osteoarthritis | TTR, Apo-A1 | Acquired | Systemic | Polyneuropathy [65] | 303 million persons w/w (80% of people > 75 years)/14.93 million new cases w/w [66,67] | 3.9% | UN/UN |
| Psoriatic Arthritis | SAA | Acquired | Systemic | Polyneuropathy [68] | 133 per 100,000 persons w/w/83 per 100,000 persons per year w/w [69] | 0.133% | UN/UN |
| Familial Mediterranean fever | SAA | Hereditary | Systemic | Polyneuropathy [22] | 100,000 persons in Turkey /UN (high among people from the eastern Mediterranean e.g., Arabs, Turks, Jews, and Armenians) [70,71] | 0.13% | UN/UN |
| Muckle–Wells syndrome | SAA | Hereditary | Systemic | Polyneuropathy [22] | Rare, MWS is one of the three clinical forms of CAPS and the prevalence of CAPS is 1–10 cases per million in France/UN [72] | 0.001% (based on max 10 per million) | UN/UN |
3.1. Familial and Acquired Amyloid Polyneuropathies
3.2. Common Acquired Diseases with Peripheral Neuropathy and Amyloid
3.2.1. Type 2 Diabetes Mellitus (T2DM)
3.2.2. Acquired Chronic Inflammatory Diseases
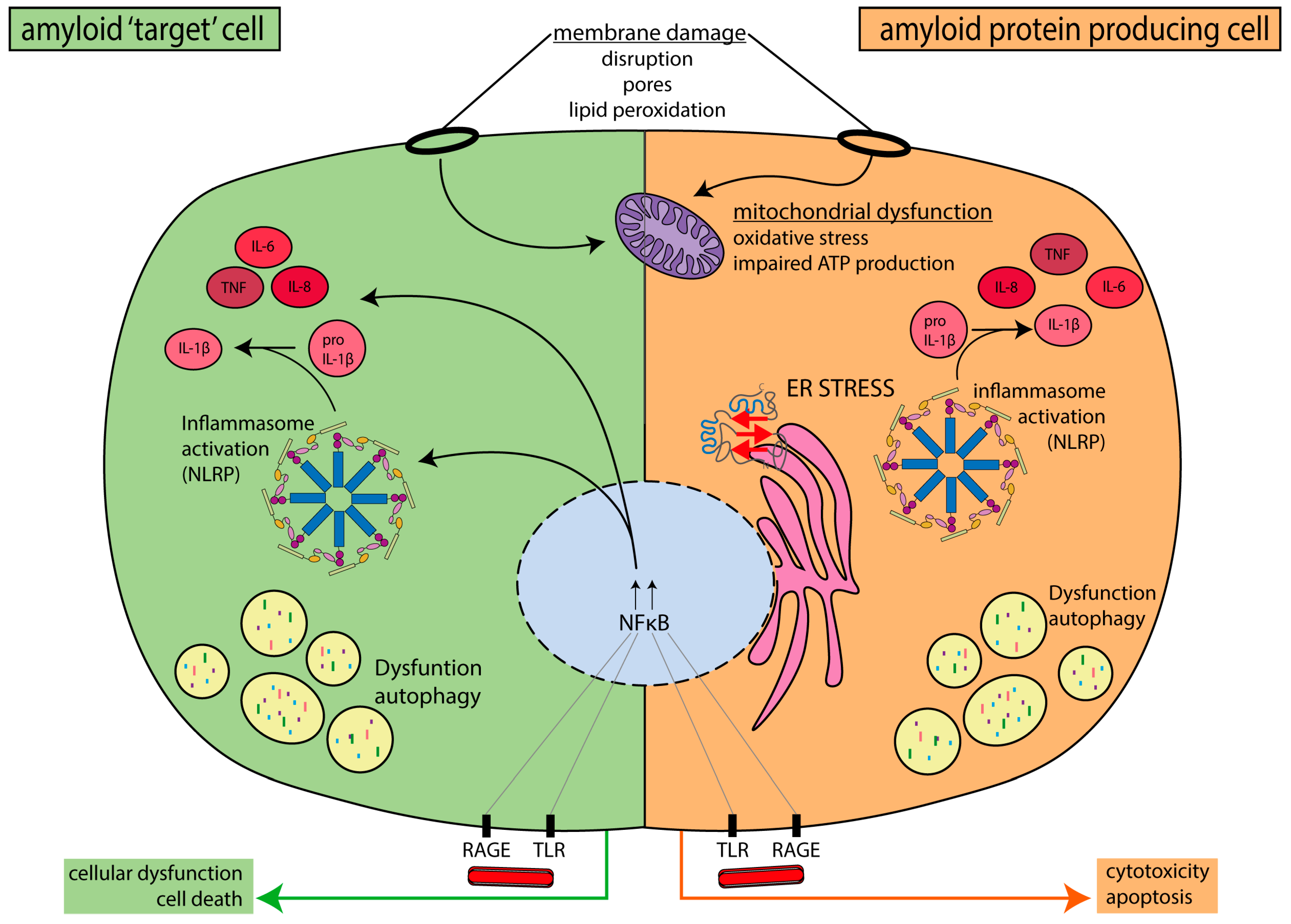
4. Mechanisms Linking Amyloid and Peripheral Neuropathy
4.1. Protein–Membrane Interactions
4.2. Endoplasmic Reticulum (ER) Stress
4.3. Mitochondrial Dysfunction
4.4. Inflammation
4.5. Macrophages and Microglia
4.6. Autophagy Impairment
4.7. Schwann Cells
4.8. Microangiopathy
5. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collins, M.P.; Dyck, P.J.B. Peripheral Nervous System Involvement. Rare Diseases of the Immune System. In Anti-Neutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis; Sinico, R., Guillevin, L., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Javed, S.; Hayat, T.; Menon, L.; Alam, U.; Malik, R.A. Diabetic peripheral neuropathy in people with type 2 diabetes: Too little too late. Diabetic Med. 2019. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Barrell, K.; Smith, A.G. Peripheral Neuropathy. Med. Clin. N. Am. 2019, 103, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Weis, J. Diseases of the peripheral nerves. Handb. Clin. Neurol. 2017, 145, 453–474. [Google Scholar] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef]
- Wechalekar, A.D.; Gillmore, J.D.; Hawkins, P.N. Systemic amyloidosis. Lancet 2016, 387, 2641–2654. [Google Scholar] [CrossRef]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am. J. Hematol. 2016, 91, 947–956. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils: Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.; Westermark, P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014, 21, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Kisilevsky, R. The relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status, 2000. Amyloid 2000, 7, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Rigacci, S. Protein folding and aggregation into amyloid: The interference by natural phenolic compounds. Int. J. Mol. Sci. 2013, 14, 12411–12457. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Vieira, M.N.; De Felice, F.G. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life 2007, 59, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V. Transthyretin familial amyloid polyneuropathy: An update. J. Neurol. 2018, 265, 976–983. [Google Scholar] [CrossRef]
- Vital, C.; Vital, A.; Bouillot-Eimer, S.; Brechenmacher, C.; Ferrer, X.; Lagueny, A. Amyloid neuropathy: A retrospective study of 35 peripheral nerve biopsies. J. Peripher. Nerv. Syst. 2004, 9, 232–241. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Klevanski, M.; Saar, M.; Muller, U.C. Roles of the amyloid precursor protein family in the peripheral nervous system. Mech. Dev. 2013, 130, 433–446. [Google Scholar] [CrossRef]
- Kanta, M.; Ehler, E.; Kohout, A.; Habalova, J.; Hanacek, R.; Vysata, O.; Brtkova, J.; Rehak, S.; Valis, M. Rare case of a localized radial nerve amyloid neuropathy. J. Clin. Neuromuscul. Dis. 2014, 16, 20–23. [Google Scholar] [CrossRef]
- Montealegre Sanchez, G.A.; Hashkes, P.J. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev. Med. Child Neurol. 2009, 51, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Real de Asua, D.; Costa, R.; Galvan, J.M.; Filigheddu, M.T.; Trujillo, D.; Cadinanos, J. Systemic AA amyloidosis: Epidemiology, diagnosis, and management. Clin. Epidemiol. 2014, 6, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Hoppener, J.W.; Ahren, B.; Lips, C.J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef]
- Jolivalt, C.G.; Calcutt, N.A.; Masliah, E. Similar pattern of peripheral neuropathy in mouse models of type 1 diabetes and Alzheimer’s disease. Neuroscience 2012, 202, 405–412. [Google Scholar] [CrossRef]
- Grambalova, Z.; Calcutt, N.A.; Masliah, E. Peripheral neuropathy in Parkinson’s disease. Neuroendocrinol. Lett. 2015, 36, 363–367. [Google Scholar]
- Shin, S.C.; Robinson-Papp, J. Amyloid neuropathies. Mt. Sinai J. Med. 2012, 79, 733–748. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013, 52, 1913–1926. [Google Scholar] [CrossRef]
- Eichner, T.; Radford, S.E. Understanding the complex mechanisms of beta2-microglobulin amyloid assembly. FEBS J. 2011, 278, 3868–3883. [Google Scholar] [CrossRef]
- Guo, J.T.; Yu, J.; Grass, D.; de Beer, F.C.; Kindy, M.S. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.P.; Al-Shawi, R.; Ellmerich, S.; Speck, I.; Aslam, S.; Hutchinson, W.L.; Mangione, P.P.; Disterer, P.; Gilbertson, J.A.; Hunt, T.; et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16115–16120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, X.; Li, J. Light chain amyloidosis: Where are the light chains from and how they play their pathogenic role? Blood Rev. 2017, 31, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H.; Ahren, B.; Sundler, F. Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am. J. Physiol. 1996, 271, E1008–E1014. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Lachmann, H.J.; Rowczenio, D.; Gilbertson, J.A.; Zeng, C.H.; Liu, Z.H.; Li, L.S.; Wechalekar, A.; Hawkins, P.N. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J. Am. Soc. Nephrol. 2009, 20, 444–451. [Google Scholar] [CrossRef]
- Cakar, A.; Durmus-Tekce, H.; Parman, Y. Familial Amyloid Polyneuropathy. Arch. Neuropsychiatry 2019, 56, 150–156. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Waddington-Cruz, M.; Ackermann, E.J.; Polydefkis, M.; Heitner, S.B.; Dyck, P.J.; Barroso, F.A.; Wang, A.K.; Berk, J.L.; Dyck, P.J.B.; Monia, B.P.; et al. Hereditary transthyretin amyloidosis: Baseline characteristics of patients in the NEURO-TTR trial. Amyloid 2018, 25, 180–188. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2018, 57, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kaku, M.; Berk, J.L. Neuropathy Associated with Systemic Amyloidosis. Semin. Neurol. 2019, 39, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Schonland, S.; Yumlu, S.; Hegenbart, U.; von Hutten, H.; Gioeva, Z.; Lohse, P.; Buttner, J.; Schmidt, H.; Rocken, C. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: Identification of three novel mutations in the APOA1 gene. J. Mol. Diagn. 2009, 11, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 1998, 5, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, V.; Rasmussen, N.; Juhl, B.R.; Gimsing, P.; Vorstrup, S. Combined bilateral submandibular and sublingual swelling, macroglossus, and carpal tunnel syndrome caused by light chain amyloidosis. Ear Nose Throat J. 1998, 77, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment. Am. J. Hematol. 2018, 93, 1169–1180. [Google Scholar] [CrossRef]
- Quock, T.P.; Yan, T.; Chang, E.; Guthrie, S.; Broder, M.S. Epidemiology of AL amyloidosis: A real-world study using US claims data. Blood Adv. 2018, 2, 1046–1053. [Google Scholar] [CrossRef]
- Cheng, R.R.; Eskandari, R.; Welsh, C.T.; Varma, A.K. A case of isolated amyloid light-chain amyloidosis of the radial nerve. J. Neurosurg. 2016, 125, 598–602. [Google Scholar] [CrossRef]
- Floege, J.; Ketteler, M. beta2-microglobulin-derived amyloidosis: An update. Kidney Int. Suppl. 2001, 78, S164–S171. [Google Scholar] [CrossRef]
- Scarpioni, R.; Ricardi, M.; Albertazzi, V.; De Amicis, S.; Rastelli, F.; Zerbini, L. Dialysis-related amyloidosis: Challenges and solutions. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 319–328. [Google Scholar] [CrossRef]
- Pinney, J.H.; Whelan, C.J.; Petrie, A.; Dungu, J.; Banypersad, S.M.; Sattianayagam, P.; Wechalekar, A.; Gibbs, S.D.; Venner, C.P.; Wassef, N.; et al. Senile systemic amyloidosis: Clinical features at presentation and outcome. J. Am. Heart Assoc. 2013, 2, e000098. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Roman-Pintos, L.M.; Villegas-Rivera, G.; Rodriguez-Carrizalez, A.D.; Miranda-Diaz, A.G.; Cardona-Munoz, E.G. Diabetic Polyneuropathy in Type 2 Diabetes Mellitus: Inflammation, Oxidative Stress, and Mitochondrial Function. J. Diabetes Res. 2016, 2016, 3425617. [Google Scholar] [CrossRef] [PubMed]
- IDF Diabetes atlas 9th edition 2019. Available online: https://www.diabetesatlas.org/en/ (accessed on 26 June 2020).
- Sun, J.; Wang, Y.; Zhang, X.; Zhu, S.; He, H. Prevalence of peripheral neuropathy in patients with diabetes: A systematic review and meta-analysis. Prim. Care Diabetes 2020. [Google Scholar] [CrossRef]
- Juster-Switlyk, K.; Smith, A.G. Updates in diabetic peripheral neuropathy. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Kaeley, N.; Ahmad, S.; Pathania, M.; Kakkar, R. Prevalence and patterns of peripheral neuropathy in patients of rheumatoid arthritis. J. Fam. Med. Prim Care 2019, 8, 22–26. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef]
- Figueroa, J.J.; Loftus, E.V., Jr.; Harmsen, W.S.; Dyck, P.J.; Klein, C.J. Peripheral neuropathy incidence in inflammatory bowel disease: A population-based study. Neurology 2013, 80, 1693–1697. [Google Scholar] [CrossRef]
- Kim, S.; Kang, S.J.; Oh, K.W.; Ahn, B.K.; Lee, H.L.; Han, D.S.; Jang, K.; Kim, Y.S. Chronic inflammatory demyelinating polyneuropathy-like neuropathy as an initial presentation of Crohn’s disease. BMC Neurol. 2015, 15, 48. [Google Scholar] [CrossRef]
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr.; Shivashankar, R.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeiseter, A.R. Updated Incidence and Prevalence of Crohn’s Disease and Ulcerative Colitis in Olmsted County, Minnesota (1970–2011). ACG 2014 Ann. Sci. Meet. 2014, 15, 10. [Google Scholar]
- Garcia-Cabo, C.; Moris, G. Peripheral neuropathy: An underreported neurologic manifestation of inflammatory bowel disease. Eur. J. Intern. Med. 2015, 26, 468–475. [Google Scholar] [CrossRef]
- Moreton, B.J.; Tew, V.; das Nair, R.; Wheeler, M.; Walsh, D.A.; Lincoln, N.B. Pain phenotype in patients with knee osteoarthritis: Classification and measurement properties of painDETECT and self-report Leeds assessment of neuropathic symptoms and signs scale in a cross-sectional study. Arthritis Care Res. Hoboken 2015, 67, 519–528. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Prevalence, Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Bijlsma, J.W. Analgesia and the patient with osteoarthritis. Am. J. Ther. 2002, 9, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswami, P.; Chapman, K.M.; Yang, M.L.; Rutkove, S.B. Psoriatic arthritis-associated polyneuropathy: A report of three cases. J. Clin. Neuromuscul. Dis. 2007, 9, 248–251. [Google Scholar] [CrossRef]
- Scotti, L.; Franchi, M.; Marchesoni, A.; Corrao, G. Prevalence and incidence of psoriatic arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2018, 48, 28–34. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Touitou, I. Familial mediterranean Fever in the world. Arthritis Rheum. 2009, 61, 1447–1453. [Google Scholar] [CrossRef]
- Ozdogan, H.; Ugurlu, S. Familial Mediterranean Fever. Presse Med. 2019, 48, e61–e76. [Google Scholar] [CrossRef]
- Tran, T.A. Muckle-Wells syndrome: Clinical perspectives. Open Access Rheumatol. 2017, 9, 123–129. [Google Scholar] [CrossRef]
- Sommer, C.; Schroder, J.M. Amyloid neuropathy: Immunocytochemical localization of intra- and extracellular immunoglobulin light chains. Acta Neuropathol. 1989, 79, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, K.; Fujimura, H.; Ueno, S.; Yoshikawa, H.; Kaido, M.; Nishimura, T.; Yorifuji, S.; Yanagihara, T. Familial amyloid polyneuropathy associated with transthyretin Gly42 mutation: A quantitative light and electron microscopic study of the peripheral nervous system. Acta Neuropathol. 1995, 90, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Du Yan, S.; Fernandes, R.; Guimaraes, A.; Stern, D.; Saraiva, M.J. Familial amyloid polyneuropathy: Receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J. Neurosci. 2001, 21, 7576–7586. [Google Scholar] [CrossRef] [PubMed]
- Kiuru-Enari, S.; Somer, H.; Seppalainen, A.M.; Notkola, I.L.; Haltia, M. Neuromuscular pathology in hereditary gelsolin amyloidosis. J. Neuropathol. Exp. Neurol. 2002, 61, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.J.; Bosch, E.P.; Dyck, P.J.; Singer, W.; Vrana, J.A.; Theis, J.D.; Dogan, A.; Klein, C.J. Amyloid-like IgM deposition neuropathy: A distinct clinico-pathologic and proteomic profiled disorder. J. Peripher. Nerv. Syst. 2012, 17, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, B.L.; Garg, C.; Vasishta, R.K.; Nada, R.; Goyal, M.K. Localised SAA amyloidosis with intra-axonal intra-myelin amyloid deposits. Pathology 2017, 49, 103–105. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Correia, S.C.; Carvalho, C.; Moreira, P.I. Mitochondria in Alzheimer’s Disease and Diabetes-Associated Neurodegeneration: License to Heal! Handb. Exp. Pharmacol. 2017, 240, 281–308. [Google Scholar]
- Moreira, P.I. Sweet Mitochondria: A Shortcut to Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1391–1401. [Google Scholar] [CrossRef]
- Pivovarov, A.S.; Calahorro, F.; Walker, R.J. Na(+)/K(+)-pump and neurotransmitter membrane receptors. Invert. Neurosci. 2018, 19, 1. [Google Scholar] [CrossRef]
- Hazenberg, B.P. Amyloidosis: A clinical overview. Rheum. Dis. Clin. N. Am. 2013, 39, 323–345. [Google Scholar] [CrossRef]
- Loavenbruck, A.J.; Singer, W.; Mauermann, M.L.; Sandroni, P.; PJ, B.D.; Gertz, M.; Klein, C.J.; Low, P.A. Transthyretin amyloid neuropathy has earlier neural involvement but better prognosis than primary amyloid counterpart: An answer to the paradox? Ann. Neurol. 2016, 80, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Said, G.; Ropert, A.; Faux, N. Length-dependent degeneration of fibers in Portuguese amyloid polyneuropathy: A clinicopathologic study. Neurology 1984, 34, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Gertz, M.A.; Kyle, R.A. Prognosis of patients with primary systemic amyloidosis who present with dominant neuropathy. Am. J. Med. 1998, 104, 232–237. [Google Scholar] [CrossRef]
- Adams, D.; Lozeron, P.; Theaudin, M.; Denier, C.; Fagniez, O.; Rerat, K.; Signate, A.; Corcia, P.; Lacroix, C. Varied patterns of inaugural light-chain (AL) amyloid polyneuropathy: A monocentric study of 24 patients. Amyloid 2011, 18, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.; Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 2017, 105, 300–320. [Google Scholar] [CrossRef]
- Pareyson, D.; Saveri, P.; Sagnelli, A.; Piscosquito, G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci. Lett. 2015, 596, 66–77. [Google Scholar] [CrossRef]
- Simmons, Z.; Specht, C.S. The neuromuscular manifestations of amyloidosis. J. Clin. Neuromuscul. Dis. 2010, 11, 145–157. [Google Scholar] [CrossRef]
- Sousa, M.M.; Saraiva, M.J. Neurodegeneration in familial amyloid polyneuropathy: From pathology to molecular signaling. Prog. Neurobiol. 2003, 71, 385–400. [Google Scholar] [CrossRef]
- Murakami, T.; Sunada, Y. Transthyretin Amyloid Neuropathy: The Schwann Cell Hypothesis. Adv. Exp. Med. Biol. 2019, 1190, 371–378. [Google Scholar]
- Sekijima, Y.; Kelly, J.W.; Ikeda, S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr. Pharm. Des. 2008, 14, 3219–3230. [Google Scholar] [CrossRef]
- Wang, A.K.; Fealey, R.D.; Gehrking, T.L.; Low, P.A. Patterns of neuropathy and autonomic failure in patients with amyloidosis. Mayo Clin. Proc. 2008, 83, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, A.; Andrade, C. Familial amyloid polyneuropathy: An electron microscope study of the peripheral nerve in five cases. I. Interstitial changes. Brain 1971, 94, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Testro, A.G.; Brennan, S.O.; Macdonell, R.A.; Hawkins, P.N.; Angus, P.W. Hereditary amyloidosis with progressive peripheral neuropathy associated with apolipoprotein AI Gly26Arg: Outcome of hepatorenal transplantation. Liver Transpl. 2007, 13, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ikeda, S.; Takahashi, M.; Kawagashira, Y.; Iijima, M.; Misumi, Y.; Ando, Y.; Ikeda, S.I.; Katsuno, M.; Sobue, G. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology 2016, 87, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.; Dogan, A.; Theis, J.D.; Vrana, J.A.; Lachmann, H.J.; Wechalekar, A.D.; Gilbertson, J.A.; Hunt, T.; Gibbs, S.D. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am. J. Pathol. 2011, 179, 1978–1987. [Google Scholar] [CrossRef]
- Paunio, T.; Kangas, H.; Kiuru, S.; Palo, J.; Peltonen, L.; Syvanen, A.C. Tissue distribution and levels of gelsolin mRNA in normal individuals and patients with gelsolin-related amyloidosis. FEBS Lett. 1997, 406, 49–55. [Google Scholar] [CrossRef]
- Burtnick, L.D.; Koepf, E.K.; Grimes, J.; Jones, E.Y.; Stuart, D.I.; McLaughlin, P.J.; Robinson, R.C. The crystal structure of plasma gelsolin: Implications for actin severing, capping, and nucleation. Cell 1997, 90, 661–670. [Google Scholar] [CrossRef]
- Bucki, R.; Levental, I.; Kulakowska, A.; Janmey, P.A. Plasma gelsolin: Function, prognostic value, and potential therapeutic use. Curr. Protein Pept. Sci. 2008, 9, 541–551. [Google Scholar] [CrossRef]
- Westberg, J.A.; Zhang, K.Z.; Andersson, L.C. Regulation of neural differentiation by normal and mutant (G654A, amyloidogenic) gelsolin. FASEB J. 1999, 13, 1621–1626. [Google Scholar] [CrossRef]
- Kyle, R.A.; Linos, A.; Beard, C.M.; Linke, R.P.; Gertz, M.A.; O’Fallon, W.M.; Kurland, L.T. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992, 79, 1817–1822. [Google Scholar] [CrossRef]
- Schonland, S.O.; Hegenbart, U.; Bochtler, T.; Mangatter, A.; Hansberg, M.; Ho, A.D.; Lohse, P.; Rocken, C. Immunohistochemistry in the classification of systemic forms of amyloidosis: A systematic investigation of 117 patients. Blood 2012, 119, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Palladini, G. Light chain amyloidosis: The heart of the problem. Haematologica 2013, 98, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, Z.; Liu, H.; Liu, D.; Gong, Z.; Qi, J.; Hu, J. Clinical characteristics and diagnosis of a rare case of systemic AL amyloidosis: A descriptive study. Oncotarget 2018, 9, 24283–24290. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, N.H. beta(2)-microglobulin: From physiology to amyloidosis. Amyloid 2009, 16, 151–173. [Google Scholar] [CrossRef]
- Haan, J.; Peters, W.G. Amyloid and peripheral nervous system disease. Clin. Neurol. Neurosurg. 1994, 96, 1–9. [Google Scholar] [CrossRef]
- O’Regan, J.; Walsh, R.; Kelly, D.; Plant, L.; Eustace, J. Neuropathy in the hemodialysis population: A review of neurophysiology referrals in a tertiary center. Ren. Fail. 2012, 34, 538–541. [Google Scholar] [CrossRef]
- Morris, A.D.; Smith, R.N.; Stone, J.R. The pathology and changing epidemiology of dialysis-related cardiac beta-2 microglobulin amyloidosis. Cardiovasc. Pathol. 2019, 42, 30–35. [Google Scholar] [CrossRef]
- Bodard, Q.; Roca, F.; Dilly, B.; Laurent, D.; Chassagne, P. Acute cardiac failure secondary to senile systemic amyloidosis. Age Ageing 2016, 45, 908–909. [Google Scholar] [CrossRef]
- Das, M.; Wilson, C.J.; Mei, X.; Wales, T.E.; Engen, J.R.; Gursky, O. Structural Stability and Local Dynamics in Disease-Causing Mutants of Human Apolipoprotein A-I: What Makes the Protein Amyloidogenic? J. Mol. Biol. 2016, 428, 449–462. [Google Scholar] [CrossRef]
- Hammarstrom, P.; Jiang, X.; Hurshman, A.R.; Powers, E.T.; Kelly, J.W. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA 2002, 99, 16427–16432. [Google Scholar] [CrossRef]
- Russo, P.; Palladini, G.; Foli, A.; Zenone Bragotti, L.; Milani, P.; Nuvolone, M.; Obici, L.; Perfetti, V.; Brugnatelli, S.; Invernizzi, R.; et al. Liver involvement as the hallmark of aggressive disease in light chain amyloidosis: Distinctive clinical features and role of light chain type in 225 patients. Amyloid 2011, 18, 92–93. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Katoh, N.; Ikeda, S. Clinical manifestations at diagnosis in Japanese patients with systemic AL amyloidosis: A retrospective study of 202 cases with a special attention to uncommon symptoms. Intern. Med. 2014, 53, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Hegenbart, U.; Milani, P.; Kimmich, C.; Foli, A.; Ho, A.D.; Vidus Rosin, M.; Albertini, R.; Moratti, R.; Merlini, G.; et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014, 124, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Wechalekar, A.; Bird, J.; Cavenagh, J.; Hawkins, S.; Kazmi, M.; Lachmann, H.J.; Hawkins, P.N.; Pratt, G.; the BCSH Committee. Guidelines on the diagnosis and investigation of AL amyloidosis. Br. J. Haematol. 2015, 168, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkanen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983, 75, 618–623. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. Lausanne 2017, 8, 343. [Google Scholar] [CrossRef]
- Cernea, S.; Dobreanu, M. Diabetes and beta cell function: From mechanisms to evaluation and clinical implications. Biochem. Med. Zagreb 2013, 23, 266–280. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef]
- Saisho, Y. beta-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109–124. [Google Scholar] [CrossRef]
- Beery, M.L.; Jacobsen, L.M.; Atkinson, M.A.; Butler, A.E.; Campbell-Thompson, M. Islet amyloidosis in a child with type 1 diabetes. Islets 2019, 11, 44–49. [Google Scholar] [CrossRef]
- Abbott, C.A.; Malik, R.A.; van Ross, E.R.; Kulkarni, J.; Boulton, A.J. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 2011, 34, 2220–2224. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Sharma, N.; Pasnoor, M.; Kluding, P.M. Painful Diabetic Peripheral Neuropathy: Presentations, Mechanisms, and Exercise Therapy. J. Diabetes Metab. 2013. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.A. Diabetic neuropathy: A focus on small fibres. Diabetes Metab. Res. Rev. 2019, 36, e3255. [Google Scholar] [CrossRef] [PubMed]
- Shillo, P.; Sloan, G.; Greig, M.; Hunt, L.; Selvarajah, D.; Elliott, J.; Gandhi, R.; Wilkinson, I.D.; Tesfaye, S. Painful and Painless Diabetic Neuropathies: What Is the Difference? Curr. Diabetes Rep. 2019, 19, 32. [Google Scholar] [CrossRef]
- Selvarajah, D.; Kar, D.; Khunti, K.; Davies, M.J.; Scott, A.R.; Walker, J.; Tesfaye, S. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019, 7, 938–948. [Google Scholar] [CrossRef]
- Premkumar, L.S.; Pabbidi, R.M. Diabetic peripheral neuropathy: Role of reactive oxygen and nitrogen species. Cell Biochem. Biophys. 2013, 67, 373–383. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef]
- Shanmugam, N.; Reddy, M.A.; Guha, M.; Natarajan, R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003, 52, 1256–1264. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Sadidi, M.; Feldman, E.L. Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2008, 9, 301–314. [Google Scholar] [CrossRef]
- Zochodne, D.W. Diabetic polyneuropathy: An update. Curr. Opin. Neurol. 2008, 21, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macri, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Lee, C.C.; Perkins, B.A.; Kayaniyil, S.; Harris, S.B.; Retnakaran, R.; Gerstein, H.C.; Zinman, B.; Hanley, A.J. Peripheral Neuropathy and Nerve Dysfunction in Individuals at High Risk for Type 2 Diabetes: The PROMISE Cohort. Diabetes Care 2015, 38, 793–800. [Google Scholar] [CrossRef]
- Grisold, A.; Callaghan, B.C.; Feldman, E.L. Mediators of diabetic neuropathy: Is hyperglycemia the only culprit? Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 103–111. [Google Scholar] [CrossRef]
- Hoppener, J.W.; Oosterwijk, C.; van Hulst, K.L.; Verbeek, J.S.; Capel, P.J.; de Koning, E.J.; Clark, A.; Jansz, H.S.; Lips, C.J. Molecular physiology of the islet amyloid polypeptide (IAPP)/amylin gene in man, rat, and transgenic mice. J. Cell Biochem. 1994, 55, 39–53. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet beta-Cells from Amylin Cytotoxicity. Cells 2018, 7, 95. [Google Scholar] [CrossRef]
- Dhanvantari, S. Unfolding the mechanisms of disease progression in permanent neonatal diabetes. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E383–E384. [Google Scholar] [CrossRef]
- Hoppener, J.W.; Oosterwijk, C.; Nieuwenhuis, M.G.; Posthuma, G.; Thijssen, J.H.; Vroom, T.M.; Ahren, B.; Lips, C.J. Extensive islet amyloid formation is induced by development of Type II diabetes mellitus and contributes to its progression: Pathogenesis of diabetes in a mouse model. Diabetologia 1999, 42, 427–434. [Google Scholar] [CrossRef]
- Chakraborty, S.; Mukherjee, B.; Basu, S. Pinpointing proline substitution to be responsible for the loss of amyloidogenesis in IAPP. Chem. Biol. Drug Des. 2013, 82, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Butler, P.C. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 2006, 47, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liu, Z.H.; Zeng, C.H.; Peng, A.; Chen, H.P.; Zhou, H.; Li, L.S. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int. 2007, 72, 213–218. [Google Scholar] [CrossRef]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef]
- Liu, M.; Verma, N.; Peng, X.; Srodulski, S.; Morris, A.; Chow, M.; Hersh, L.B.; Chen, J.; Zhu, H.; Netea, M.G.; et al. Hyperamylinemia Increases IL-1beta Synthesis in the Heart via Peroxidative Sarcolemmal Injury. Diabetes 2016, 65, 2772–2783. [Google Scholar] [CrossRef]
- Mulder, H.; Leckstrom, A.; Uddman, R.; Ekblad, E.; Westermark, P.; Sundler, F. Islet amyloid polypeptide (amylin) is expressed in sensory neurons. J. Neurosci. 1995, 15, 7625–7632. [Google Scholar] [CrossRef]
- Mulder, H.; Zhang, Y.; Danielsen, N.; Sundler, F. Islet amyloid polypeptide and calcitonin gene-related peptide expression are upregulated in lumbar dorsal root ganglia after unilateral adjuvant-induced inflammation in the rat paw. Brain Res. Mol. Brain Res. 1997, 50, 127–135. [Google Scholar] [CrossRef]
- Mulder, H.; Zhang, Y.; Danielsen, N.; Sundler, F. Islet amyloid polypeptide and calcitonin gene-related peptide expression are down-regulated in dorsal root ganglia upon sciatic nerve transection. Brain Res. Mol. Brain Res. 1997, 47, 322–330. [Google Scholar] [CrossRef]
- Gebre-Medhin, S.; Mulder, H.; Pekny, M.; Westermark, G.; Tornell, J.; Westermark, P.; Sundler, F.; Ahren, B. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem. Biophys. Res. Commun. 1998, 250, 271–277. [Google Scholar] [CrossRef]
- Mulder, H.; Jongsma, H.; Zhang, Y.; Gebre-Medhin, S.; Sundler, F.; Danielsen, N. Pituitary adenylate cyclase-activating polypeptide and islet amyloid polypeptide in primary sensory neurons: Functional implications from plasticity in expression on nerve injury and inflammation. Mol. Neurobiol. 1999, 19, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.S.; Castro-Lopes, J.M.; Neto, F.L.; Potes, C.S. Amylin, a peptide expressed by nociceptors, modulates chronic neuropathic pain. Eur. J. Pain 2019, 23, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Gebre-Medhin, S.; Mulder, H.; Zhang, Y.; Sundler, F.; Betsholtz, C. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol. Brain Res. 1998, 63, 180–183. [Google Scholar] [CrossRef]
- Asiri, M.M.H.; Versteeg, S.; Höppener, J.W.M.; Eijkelkamp, N. Human IAPP mediates painful diabetic peripheral neuropathy. In Proceedings of the The 11th Congress of the European Pain Federation EFIC, Valencia, Spain, 4–7 September 2019. [Google Scholar]
- Hoppener, J.W.; Verbeek, J.S.; de Koning, E.J.; Oosterwijk, C.; van Hulst, K.L.; Visser-Vernooy, H.J.; Hofhuis, F.M.; van Gaalen, S.; Berends, M.J.; Hackeng, W.H.; et al. Chronic overproduction of islet amyloid polypeptide/amylin in transgenic mice: Lysosomal localization of human islet amyloid polypeptide and lack of marked hyperglycaemia or hyperinsulinaemia. Diabetologia 1993, 36, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Potes, C.S.; Pestana, A.C.; Pontes, M.; Caramelo, A.S.; Neto, F.L. Amylin modulates the formalin-induced tonic pain behaviours in rats. Eur. J. Pain 2016, 20, 1741–1752. [Google Scholar] [CrossRef]
- Verma, N.; Ly, H.; Liu, M.; Chen, J.; Zhu, H.; Chow, M.; Hersh, L.B.; Despa, F. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1beta Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J. Alzheimers Dis. 2016, 53, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Westermark, G.T.; Fandrich, M.; Westermark, P. AA amyloidosis: Pathogenesis and targeted therapy. Annu. Rev. Pathol. 2015, 10, 321–344. [Google Scholar] [CrossRef]
- Papa, R.; Lachmann, H.J. Secondary, AA, Amyloidosis. Rheum. Dis. Clin. N. Am. 2018, 44, 585–603. [Google Scholar] [CrossRef]
- Agarwal, V.; Lachmann, H.J. A clinical, electrophysiological, and pathological study of neuropathy in rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 841–844. [Google Scholar] [CrossRef]
- Akasaki, Y.; Reixach, N.; Matsuzaki, T.; Alvarez-Garcia, O.; Olmer, M.; Iwamoto, Y.; Buxbaum, J.N.; Lotz, M.K. Transthyretin deposition in articular cartilage: A novel mechanism in the pathogenesis of osteoarthritis. Arthritis Rheumatol. 2015, 67, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, A.; Ueda, M.; Sueyoshi, T.; Nakamura, E.; Tasaki, M.; Suenaga, G.; Motokawa, H.; Toyoshima, R.; Kinoshita, Y.; Misumi, Y.; et al. Knee osteoarthritis associated with different kinds of amyloid deposits and the impact of aging on type of amyloid. Amyloid 2016, 23, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bergis, M.; Dega, H.; Planquois, V.; Benichou, O.; Dubertret, L. Amyloidosis complicating psoriatic arthritis. Ann. Dermatol. Venereol. 2003, 130, 1039–1042. [Google Scholar] [PubMed]
- Basturk, T.; Ozagari, A.; Ozturk, T.; Kusaslan, R.; Unsal, A. Crohn’s disease and secondary amyloidosis: Early complication? A case report and review of the literature. J. Ren. Care 2009, 35, 147–150. [Google Scholar] [CrossRef]
- Pukitis, A.; Zake, T.; Groma, V.; Ostrovskis, E.; Skuja, S.; Pokrotnieks, J. Effect of infliximab induction therapy on secondary systemic amyloidosis associated with Crohn’s disease: Case report and review of the literature. J. Gastrointestin Liver Dis. 2013, 22, 333–336. [Google Scholar]
- Tada, Y.; Ishihara, S.; Ito, T.; Matsui, K.; Sonoyama, H.; Oka, A.; Kusunoki, R.; Fukuba, N.; Mishima, Y.; Oshima, N.; et al. Successful use of maintenance infliximab for nephropathy in a patient with secondary amyloidosis complicating Crohn’s disease. Intern. Med. 2013, 52, 1899–1902. [Google Scholar] [CrossRef]
- Vahdat Shariat Panahi, A.; Hultman, P.; Ollinger, K.; Westermark, G.T.; Lundmark, K. Lipid membranes accelerate amyloid formation in the mouse model of AA amyloidosis. Amyloid 2019, 26, 34–44. [Google Scholar] [CrossRef]
- Tanaka, M.; Nishimura, A.; Takeshita, H.; Takase, H.; Yamada, T.; Mukai, T. Effect of lipid environment on amyloid fibril formation of human serum amyloid A. Chem. Phys. Lipids 2017, 202, 6–12. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Fandrich, M.; Gursky, O. Serum amyloid A sequesters diverse phospholipids and their hydrolytic products, hampering fibril formation and proteolysis in a lipid-dependent manner. Chem. Commun. Camb. 2018, 54, 3532–3535. [Google Scholar] [CrossRef]
- Morgado, I.; Garvey, M. Lipids in Amyloid-beta Processing, Aggregation, and Toxicity. Adv. Exp. Med. Biol. 2015, 855, 67–94. [Google Scholar]
- Qiang, W.; Yau, W.M.; Schulte, J. Fibrillation of beta amyloid peptides in the presence of phospholipid bilayers and the consequent membrane disruption. Biochim. Biophys. Acta 2015, 1848, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ren, B.; Liu, Y.; Liang, G.; Sun, Y.; Xu, L.; Zheng, J. Membrane Interactions of hIAPP Monomer and Oligomer with Lipid Membranes by Molecular Dynamics Simulations. ACS Chem. Neurosci. 2017, 8, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.M.; Mok, Y.F.; Howlett, G.J.; Griffin, M.D. The Role of Lipid in Misfolding and Amyloid Fibril Formation by Apolipoprotein C-II. Adv. Exp. Med. Biol. 2015, 855, 157–174. [Google Scholar] [PubMed]
- Bram, Y.; Frydman-Marom, A.; Yanai, I.; Gilead, S.; Shaltiel-Karyo, R.; Amdursky, N.; Gazit, E. Apoptosis induced by islet amyloid polypeptide soluble oligomers is neutralized by diabetes-associated specific antibodies. Sci. Rep. 2014, 4, 4267. [Google Scholar] [CrossRef]
- Kumar, S.; Birol, M.; Miranker, A.D. Foldamer scaffolds suggest distinct structures are associated with alternative gains-of-function in a preamyloid toxin. Chem. Commun. Camb. 2016, 52, 6391–6394. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Gursky, O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. USA 2017, 114, E6507–E6515. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid growth and membrane damage: Current themes and emerging perspectives from theory and experiments on Abeta and hIAPP. Biochim. Biophys. Acta Biomembr. 2018. [Google Scholar] [CrossRef]
- Rawat, A.; Langen, R.; Varkey, J. Membranes as modulators of amyloid protein misfolding and target of toxicity. Biochim. Biophys. Acta Biomembr. 2018. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef]
- Kagan, B.L.; Thundimadathil, J. Amyloid peptide pores and the beta sheet conformation. Adv. Exp. Med. Biol. 2010, 677, 150–167. [Google Scholar] [PubMed]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—Protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar] [PubMed]
- Gurlo, T.; Ryazantsev, S.; Huang, C.J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for proteotoxicity in beta cells in type 2 diabetes: Toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 2010, 176, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and Processing of the Amyloid-beta Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimers Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ueda, Y.; Nakajima, A.; Mitsuyama, Y. EPR evidence of hydroxyl radical generation as an initiator of lipid peroxidation in amyloid beta-protein-stimulated PC12 cells. Brain Res. 2004, 1025, 29–34. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Gunn, A.P.; Wong, B.X.; Johanssen, T.; Griffith, J.C.; Masters, C.L.; Bush, A.I.; Barnham, K.J.; Duce, J.A.; Cherny, R.A. Amyloid-beta Peptide Abeta3pE-42 Induces Lipid Peroxidation, Membrane Permeabilization, and Calcium Influx in Neurons. J. Biol. Chem. 2016, 291, 6134–6145. [Google Scholar] [CrossRef]
- Hassler, S.N.; Johnson, K.M.; Hulsebosch, C.E. Reactive oxygen species and lipid peroxidation inhibitors reduce mechanical sensitivity in a chronic neuropathic pain model of spinal cord injury in rats. J. Neurochem. 2014, 131, 413–417. [Google Scholar] [CrossRef]
- Chadwick, S.R.; Lajoie, P. Endoplasmic Reticulum Stress Coping Mechanisms and Lifespan Regulation in Health and Diseases. Front. Cell Dev. Biol. 2019, 7, 84. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Genereux, J.C.; Wiseman, R.L. Endoplasmic reticulum quality control and systemic amyloid disease: Impacting protein stability from the inside out. IUBMB Life 2015, 67, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.F.; Cerca, F.; Santos, S.D.; Saraiva, M.J. Endoplasmic reticulum stress associated with extracellular aggregates. Evidence from transthyretin deposition in familial amyloid polyneuropathy. J. Biol. Chem. 2006, 281, 21998–22003. [Google Scholar] [CrossRef]
- Casas, S.; Gomis, R.; Gribble, F.M.; Altirriba, J.; Knuutila, S.; Novials, A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes 2007, 56, 2284–2294. [Google Scholar] [CrossRef]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef]
- Matveyenko, A.V.; Gurlo, T.; Daval, M.; Butler, A.E.; Butler, P.C. Successful versus failed adaptation to high-fat diet-induced insulin resistance: The role of IAPP-induced beta-cell endoplasmic reticulum stress. Diabetes 2009, 58, 906–916. [Google Scholar] [CrossRef]
- Cadavez, L.; Montane, J.; Alcarraz-Vizan, G.; Visa, M.; Vidal-Fabrega, L.; Servitja, J.M.; Novials, A. Chaperones ameliorate beta cell dysfunction associated with human islet amyloid polypeptide overexpression. PLoS ONE 2014, 9, e101797. [Google Scholar] [CrossRef]
- Law, C.J.; Ashcroft, H.A.; Zheng, W.; Sexton, J.Z. Assay development and multivariate scoring for high-content discovery of chemoprotectants of endoplasmic-reticulum-stress-mediated amylin-induced cytotoxicity in pancreatic beta cells. Assay Drug Dev. Technol. 2014, 12, 375–384. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, H.K.; Eum, W.S.; Park, J.; Choi, S.Y.; Kwon, H.Y. Tat-biliverdin reductase A protects INS-1 cells from human islet amyloid polypeptide-induced cytotoxicity by alleviating oxidative stress and ER stress. Cell Biol. Int. 2017, 41, 514–524. [Google Scholar] [CrossRef]
- He, Y.M.; Zhang, Q.; Zheng, M.; Fan, Z.H.; Li, Y.H.; Zhang, D.; Zhang, Z.; Yuan, S.S.; Wang, Y.Y.; Zhou, P.; et al. Protective effects of a G. lucidum proteoglycan on INS-1 cells against IAPP-induced apoptosis via attenuating endoplasmic reticulum stress and modulating CHOP/JNK pathways. Int. J. Biol. Macromol. 2018, 106, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Ito, K.; Teraoka, R.; Lambert, M.P.; Klein, W.L.; Mori, H. The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 2009, 174, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef]
- Lupachyk, S.; Watcho, P.; Obrosov, A.A.; Stavniichuk, R.; Obrosova, I.G. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp. Neurol. 2013, 247, 342–348. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Sakowski, S.A.; Feldman, E.L. ER stress in diabetic peripheral neuropathy: A new therapeutic target. Antioxid. Redox. Signal. 2014, 21, 621–633. [Google Scholar]
- Inceoglu, B.; Bettaieb, A.; Trindade da Silva, C.A.; Lee, K.S.; Haj, F.G.; Hammock, B.D. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef]
- Lupachyk, S.; Watcho, P.; Stavniichuk, R.; Shevalye, H.; Obrosova, I.G. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes 2013, 62, 944–952. [Google Scholar] [CrossRef]
- Valenzuela, V.; Onate, M.; Hetz, C.; Court, F.A. Injury to the nervous system: A look into the ER. Brain Res. 2016, 1648, 617–625. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid. Med. Cell Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef] [PubMed]
- Rumora, A.E.; Savelieff, M.G.; Sakowski, S.A.; Feldman, E.L. Disorders of mitochondrial dynamics in peripheral neuropathy: Clues from hereditary neuropathy and diabetes. Int. Rev. Neurobiol. 2019, 145, 127–176. [Google Scholar] [PubMed]
- Trecarichi, A.; Flatters, S.J.L. Mitochondrial dysfunction in the pathogenesis of chemotherapy-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 83–126. [Google Scholar]
- Olsson, M.; Hellman, U.; Plante-Bordeneuve, V.; Jonasson, J.; Lang, K.; Suhr, O.B. Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin. Genet. 2009, 75, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bonaiti, B.; Olsson, M.; Hellman, U.; Suhr, O.; Bonaiti-Pellie, C.; Plante-Bordeneuve, V. TTR familial amyloid polyneuropathy: Does a mitochondrial polymorphism entirely explain the parent-of-origin difference in penetrance? Eur. J. Hum. Genet. 2010, 18, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Akhter, F.; Chen, D.; Yan, S.F.; Yan, S.S. Mitochondrial Perturbation in Alzheimer’s Disease and Diabetes. Prog. Mol. Biol. Transl. Sci. 2017, 146, 341–361. [Google Scholar]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Lim, Y.A.; Rhein, V.; Baysang, G.; Meier, F.; Poljak, A.; Raftery, M.J.; Guilhaus, M.; Ittner, L.M.; Eckert, A.; Gotz, J. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics 2010, 10, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Kumar, S.; Rhoades, E.; Miranker, A.D. Conformational switching within dynamic oligomers underpins toxic gain-of-function by diabetes-associated amyloid. Nat. Commun. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca(2+) Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca(2+) Remodeling. Front. Cell Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schutz, O.; et al. alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol. 2019, 19, 146. [Google Scholar] [CrossRef]
- Abedini, A.; Derk, J.; Schmidt, A.M. The receptor for advanced glycation endproducts is a mediator of toxicity by IAPP and other proteotoxic aggregates: Establishing and exploiting common ground for novel amyloidosis therapies. Protein Sci. 2018, 27, 1166–1180. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflamm. 2013, 2013, 403460. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Sousa, M.M.; Yan, S.D.; Stern, D.; Saraiva, M.J. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.L.; Yan, S.F.; Wendt, T.; Hans, D.; Pachydaki, S.; Bucciarelli, L.G.; Adebayo, A.; Qu, W.; Lu, Y.; Kostov, K.; et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004, 18, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Smith, D.R.; Tessler, L.; Mateo, A.R.; Martens, C.; Schartner, E.; Van der Ploeg, R.; Toth, C.; Zochodne, D.W.; Fernyhough, P. Receptor for advanced glycation end-products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp. Neurol. 2013, 249, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Brederson, J.D.; Strakhova, M.; Mills, C.; Barlow, E.; Meyer, A.; Nimmrich, V.; Leddy, M.; Simler, G.; Schmidt, M.; Jarvis, M.; et al. A monoclonal antibody against the receptor for advanced glycation end products attenuates inflammatory and neuropathic pain in the mouse. Eur. J. Pain 2016, 20, 607–614. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef]
- Sommer, C.; Leinders, M.; Uceyler, N. Inflammation in the pathophysiology of neuropathic pain. Pain 2018, 159, 595–602. [Google Scholar] [CrossRef]
- Safieh-Garabedian, B.; Nomikos, M.; Saade, N. Targeting inflammatory components in neuropathic pain: The analgesic effect of thymulin related peptide. Neurosci. Lett. 2019, 702, 61–65. [Google Scholar] [CrossRef]
- Raoof, R.; Willemen, H.; Eijkelkamp, N. Divergent roles of immune cells and their mediators in pain. Rheumatology 2018, 57, 429–440. [Google Scholar] [CrossRef]
- Coll, R.C.; O’Neill, L.; Schroder, K. Questions and controversies in innate immune research: What is the physiological role of NLRP3? Cell Death Discov. 2016, 2, 16019. [Google Scholar] [CrossRef]
- Migita, K.; Izumi, Y.; Jiuchi, Y.; Kozuru, H.; Kawahara, C.; Nakamura, M.; Nakamura, T.; Agematsu, K.; Masumoto, J.; Yasunami, M.; et al. Serum amyloid A induces NLRP-3-mediated IL-1beta secretion in neutrophils. PLoS ONE 2014, 9, e96703. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Liu, S.; Yi, X.; Zhang, S.; Ding, Y. Serum amyloid A induces interleukin-1beta secretion from keratinocytes via the NACHT, LRR and PYD domains-containing protein 3 inflammasome. Clin. Exp. Immunol. 2015, 179, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Song, L.T.; Lai, W.; Li, J.S.; Mu, Y.Z.; Li, C.Y.; Jiang, S.Y. The interaction between serum amyloid A and Toll-like receptor 2 pathway regulates inflammatory cytokine secretion in human gingival fibroblasts. J. Periodontol. 2020, 91, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic beta cell proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes 2014, 63, 1698–1711. [Google Scholar] [CrossRef]
- Park, Y.J.; Warnock, G.L.; Ao, Z.; Safikhan, N.; Meloche, M.; Asadi, A.; Kieffer, T.J.; Marzban, L. Dual role of interleukin-1beta in islet amyloid formation and its beta-cell toxicity: Implications for type 2 diabetes and islet transplantation. Diabetes Obes. Metab. 2017, 19, 682–694. [Google Scholar] [CrossRef]
- Mu, Z.P.; Wang, Y.G.; Li, C.Q.; Lv, W.S.; Wang, B.; Jing, Z.H.; Song, X.J.; Lun, Y.; Qiu, M.Y.; Ma, X.L. Association Between Tumor Necrosis Factor-alpha and Diabetic Peripheral Neuropathy in Patients with Type 2 Diabetes: A Meta-Analysis. Mol. Neurobiol. 2017, 54, 983–996. [Google Scholar] [CrossRef]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The Role of Neuronal NLRP1 Inflammasome in Alzheimer’s Disease: Bringing Neurons into the Neuroinflammation Game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef] [PubMed]
- Nan, K.; Han, Y.; Fang, Q.; Huang, C.; Yu, L.; Ge, W.; Xiang, F.; Tao, Y.X.; Cao, H.; Li, J. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-kappaB signaling in primary hippocampal neurons induced by Abeta25-35. Int. Immunopharmacol. 2019, 67, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lathia, J.D.; Selvaraj, P.K.; Jo, D.G.; Mughal, M.R.; Cheng, A.; Siler, D.A.; Markesbery, W.R.; Arumugam, T.V.; Mattson, M.P. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 2008, 213, 114–121. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Moyse, E.; Haddad, M.; Benlabiod, C.; Ramassamy, C.; Krantic, S. Common Pathological Mechanisms and Risk Factors for Alzheimer’s Disease and Type-2 Diabetes: Focus on Inflammation. Curr. Alzheimer Res. 2019, 16, 986–1006. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimers Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef]
- Man, L.L.; Liu, F.; Wang, Y.J.; Song, H.H.; Xu, H.B.; Zhu, Z.W.; Zhang, Q.; Wang, Y.J. The HMGB1 signaling pathway activates the inflammatory response in Schwann cells. Neural Regen. Res. 2015, 10, 1706–1712. [Google Scholar]
- Salvador, B.; Arranz, A.; Francisco, S.; Cordoba, L.; Punzon, C.; Llamas, M.A.; Fresno, M. Modulation of endothelial function by Toll like receptors. Pharmacol. Res. 2016, 108, 46–56. [Google Scholar] [CrossRef]
- Jiang, T.; Jiang, D.; Zhang, L.; Ding, M.; Zhou, H. Anagliptin ameliorates high glucose-induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol. Immunol. 2019, 107, 54–60. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Z.Z.; Zhang, Y.M.; Kong, L.; Xiao, W.F.; Ma, T.F.; Liu, Y.W. Thrombin/PAR-1 activation induces endothelial damages via NLRP1 inflammasome in gestational diabetes. Biochem. Pharmacol. 2020, 175, 113849. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Ando, Y.; Goncalves, N.P.; Saraiva, M.J. Fibroblasts endocytose and degrade transthyretin aggregates in transthyretin-related amyloidosis. Lab. Investig. 2013, 93, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Fella, E.; Sokratous, K.; Papacharalambous, R.; Kyriacou, K.; Phillips, J.; Sanderson, S.; Panayiotou, E.; Kyriakides, T. Pharmacological Stimulation of Phagocytosis Enhances Amyloid Plaque Clearance; Evidence from a Transgenic Mouse Model of ATTR Neuropathy. Front. Mol. Neurosci. 2017, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, E.; Fella, E.; Papacharalambous, R.; Malas, S.; Saraiva, M.J.; Kyriakides, T. C1q ablation exacerbates amyloid deposition: A study in a transgenic mouse model of ATTRV30M amyloid neuropathy. PLoS ONE 2017, 12, e0175767. [Google Scholar] [CrossRef]
- Garcia-Garcia, M.A.R.; Argilés, À.; Gouin-Charnet, A.; Durfort, M.; Garcia-Valero, J.; Mourad, G. Impaired lysosomal processing of beta2-microglobulin by infiltrating macrophages in dialysis amyloidosis. Kidney Int. 1999, 55, 899–906. [Google Scholar] [CrossRef]
- Okoshi, T.; Yamaguchi, I.; Ozawa, D.; Hasegawa, K.; Naiki, H. Endocytosed 2-Microglobulin Amyloid Fibrils Induce Necrosis and Apoptosis of Rabbit Synovial Fibroblasts by Disrupting Endosomal/Lysosomal Membranes: A Novel Mechanism on the Cytotoxicity of Amyloid Fibrils. PLoS ONE 2015, 10, e0139330. [Google Scholar] [CrossRef]
- Suenaga, G.; Ikeda, T.; Komohara, Y.; Takamatsu, K.; Kakuma, T.; Tasaki, M.; Misumi, Y.; Ueda, M.; Ito, T.; Senju, S.; et al. Involvement of Macrophages in the Pathogenesis of Familial Amyloid Polyneuropathy and Efficacy of Human iPS Cell-Derived Macrophages in Its Treatment. PLoS ONE 2016, 11, e0163944. [Google Scholar] [CrossRef]
- Jabaut, J.; Ather, J.L.; Taracanova, A.; Poynter, M.E.; Ckless, K. Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1beta secretion in association with alterations in cellular redox and energy status. Free Radic. Biol. Med. 2013, 60, 233–245. [Google Scholar] [CrossRef]
- Niemi, K.; Teirila, L.; Lappalainen, J.; Rajamaki, K.; Baumann, M.H.; Oorni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Nackiewicz, D.; Dan, M.; Ehses, J.A. Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol. Cell Biol. 2014, 92, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.J.; Tang, L.; Zhao, X.P.; Xu, J.M.; Xiao, Y.; Li, H. Infiltration of Blood-Derived Macrophages Contributes to the Development of Diabetic Neuropathy. J. Immunol. Res. 2019, 2019, 7597382. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Vazquez, P.A.; Grosick, R.L.; Moracho-Vilrriales, C.; Ward, E.; Threatt, T.; Romero-Sandoval, E.A. Cytokine production capabilities of human primary monocyte-derived macrophages from patients with diabetes mellitus type 2 with and without diabetic peripheral neuropathy. J. Pain Res. 2019, 12, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Couture, R.; Hong, Y. Activated microglia in the spinal cord underlies diabetic neuropathic pain. Eur. J. Pharmacol. 2014, 728, 59–66. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Ledo, J.H.; Barbosa, G.; Sobrinho, M.; Diniz, L.; Fonseca, A.C.; Gomes, F.; Romao, L.; Lima, F.R.; Palhano, F.L.; et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 2013, 4, e789. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, H.; Taheri, S.; Mondy, W.; Bonilha, L.; Magwood, G.S.; Lackland, D.; Adams, R.J.; Kindy, M.S. Serum Amyloid A-Mediated Inflammasome Activation of Microglial Cells in Cerebral Ischemia. J. Neurosci. 2019, 39, 9465–9476. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, H.; Hamel, K.A.; Morvan, M.G.; Yu, S.; Leff, J.; Guan, Z.; Braz, J.M.; Basbaum, A.I. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat. Commun. 2020, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M. Microglia in the spinal cord and neuropathic pain. J. Diabetes Investig. 2016, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Rajchgot, T.; Thomas, S.C.; Wang, J.C.; Ahmadi, M.; Balood, M.; Crosson, T.; Dias, J.P.; Couture, R.; Claing, A.; Talbot, S. Neurons and Microglia; A Sickly-Sweet Duo in Diabetic Pain Neuropathy. Front. Neurosci. 2019, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Y.; Cao, L.; Xiong, R.; Zhang, B.; Wu, L.; Zhao, Z.; Chen, S.D. Curcumin could reduce the monomer of TTR with Tyr114Cys mutation via autophagy in cell model of familial amyloid polyneuropathy. Drug Des. Dev. Ther. 2014, 8, 2121–2128. [Google Scholar]
- Teixeira, C.A.; Almeida Mdo, R.; Saraiva, M.J. Impairment of autophagy by TTR V30M aggregates: In vivo reversal by TUDCA and curcumin. Clin. Sci. Lond. 2016, 130, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, M.; Ju, Y.; Li, A.; Sun, X. Autophagy dysfunction in neuropathic pain. Neuropeptides 2019, 75, 41–48. [Google Scholar] [CrossRef]
- Yin, Y.; Yi, M.H.; Kim, D.W. Impaired Autophagy of GABAergic Interneurons in Neuropathic Pain. Pain Res. Manag. 2018, 2018, 9185368. [Google Scholar] [CrossRef]
- Haidar, M.; Timmerman, V. Autophagy as an Emerging Common Pathomechanism in Inherited Peripheral Neuropathies. Front. Mol. Neurosci. 2017, 10, 143. [Google Scholar] [CrossRef]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef]
- Osorio, J. Diabetes: Protective role of autophagy in pancreatic beta cells. Nat. Rev. Endocrinol. 2014, 10, 575. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.; Park, K.; Lee, M.S. beta-cell autophagy: Mechanism and role in beta-cell dysfunction. Mol. Metab. 2019, 27, S92–S103. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Resende, R.; Moreira, P.I.; Pereira, C.M. Alzheimer’s disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol. 2015, 34, 261–273. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Hernandez, N.; Sproul, A.A.; Yu, W.H. Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 2019, 697, 49–58. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, R.; Bokil, N.J.; Sweet, M.J. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: A ticking time bomb in ageing. Ageing Res. Rev. 2015, 24, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.; Martins, R.; Bharadwaj, P. Autophagy Modulation as a Treatment of Amyloid Diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [PubMed]
- Said, G. Familial amyloid polyneuropathy: Mechanisms leading to nerve degeneration. Amyloid 2003, 10, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Lozeron, P.; Mariani, L.L.; Dodet, P.; Beaudonnet, G.; Theaudin, M.; Adam, C.; Arnulf, B.; Adams, D. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology 2018, 91, e143–e152. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Fei, Y.; Su, W.; Chen, G. Emerging Role of Schwann Cells in Neuropathic Pain: Receptors, Glial Mediators and Myelination. Front. Cell Neurosci. 2019, 13, 116. [Google Scholar] [CrossRef]
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimaraes, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: Evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 2001, 159, 1993–2000. [Google Scholar] [CrossRef]
- Murakami, T.; Sango, K.; Watabe, K.; Niimi, N.; Takaku, S.; Li, Z.; Yamamura, K.; Sunada, Y. Schwann cells contribute to neurodegeneration in transthyretin amyloidosis. J. Neurochem. 2015, 134, 66–74. [Google Scholar] [CrossRef]
- Jang, S.Y.; Shin, Y.K.; Lee, H.Y.; Park, J.Y.; Suh, D.J.; Kim, J.K.; Bae, Y.S.; Park, H.T. Local production of serum amyloid a is implicated in the induction of macrophage chemoattractants in Schwann cells during wallerian degeneration of peripheral nerves. Glia 2012, 60, 1619–1628. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines 2019, 7, 11. [Google Scholar] [CrossRef]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, M.; Mourad, G.; Durfort, M.; Garcia-Valero, J.; Argiles, A. Vascular involvement and cell damage in experimental AA and clinical beta(2)-microglobulin amyloidosis. Nephrol. Dial. Transplant. 2002, 17, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Belmokhtar, K.; Robert, T.; Ortillon, J.; Braconnier, A.; Vuiblet, V.; Boulagnon-Rombi, C.; Diebold, M.D.; Pietrement, C.; Schmidt, A.M.; Rieu, P.; et al. Signaling of Serum Amyloid A Through Receptor for Advanced Glycation End Products as a Possible Mechanism for Uremia-Related Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, J.; Wang, S. Serum Amyloid A Induces a Vascular Smooth Muscle Cell Phenotype Switch through the p38 MAPK Signaling Pathway. Biomed. Res. Int. 2017, 2017, 4941379. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Aino, M.; Kobayashi, S.I.; Okada, K.; Ohno, T.; Kikuchi, T.; Hayashi, J.I.; Yamamoto, G.; Hasegawa, Y.; Mitani, A. Serum Amyloid A Promotes E-Selectin Expression via Toll-Like Receptor 2 in Human Aortic Endothelial Cells. Mediators Inflamm. 2016, 2016, 7150509. [Google Scholar] [CrossRef] [PubMed]
- Chami, B.; Barrie, N.; Cai, X.; Wang, X.; Paul, M.; Morton-Chandra, R.; Sharland, A.; Dennis, J.M.; Freedman, S.B.; Witting, P.K. Serum amyloid A receptor blockade and incorporation into high-density lipoprotein modulates its pro-inflammatory and pro-thrombotic activities on vascular endothelial cells. Int. J. Mol. Sci. 2015, 16, 11101–11124. [Google Scholar] [CrossRef]
- Brissova, M.; Shostak, A.; Fligner, C.L.; Revetta, F.L.; Washington, M.K.; Powers, A.C.; Hull, R.L. Human Islets Have Fewer Blood Vessels than Mouse Islets and the Density of Islet Vascular Structures Is Increased in Type 2 Diabetes. J. Histochem. Cytochem. 2015, 63, 637–645. [Google Scholar] [CrossRef]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Lastra, G.; Patel, K.; Wasekar, C.; Manrique, C.M.; Ozerdem, U.; Stas, S.; Sowers, J.R. Ultrastructure of islet microcirculation, pericytes and the islet exocrine interface in the HIP rat model of diabetes. Exp. Biol. Med. Maywood 2008, 233, 1109–1123. [Google Scholar] [CrossRef]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef]
- Ly, H.; Verma, N.; Wu, F.; Liu, M.; Saatman, K.E.; Nelson, P.T.; Slevin, J.T.; Goldstein, L.B.; Biessels, G.J.; Despa, F. Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann. Neurol. 2017, 82, 208–222. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L. Increased permeability across the blood-nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy. Proc. Natl. Acad. Sci. USA 1992, 89, 2218–2222. [Google Scholar] [CrossRef] [PubMed]
- Mizisin, A.P.; Weerasuriya, A. Homeostatic regulation of the endoneurial microenvironment during development, aging and in response to trauma, disease and toxic insult. Acta Neuropathol. 2011, 121, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Richner, M.; Ferreira, N.; Dudele, A.; Jensen, T.S.; Vaegter, C.B.; Goncalves, N.P. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front. Neurosci. 2018, 12, 1038. [Google Scholar] [CrossRef]
- Bengoechea, T.G.; Chen, Z.; O’Leary, D.A.; Masliah, E.; Lee, K.F. p75 reduces beta-amyloid-induced sympathetic innervation deficits in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2009, 106, 7870–7875. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albariqi, M.M.H.; Engelsman, S.; Eijkelkamp, N.; Höppener, J.W.M. Amyloid Proteins and Peripheral Neuropathy. Cells 2020, 9, 1553. https://doi.org/10.3390/cells9061553
Albariqi MMH, Engelsman S, Eijkelkamp N, Höppener JWM. Amyloid Proteins and Peripheral Neuropathy. Cells. 2020; 9(6):1553. https://doi.org/10.3390/cells9061553
Chicago/Turabian StyleAlbariqi, Mohammed M. H., Sjoukje Engelsman, Niels Eijkelkamp, and Jo W. M. Höppener. 2020. "Amyloid Proteins and Peripheral Neuropathy" Cells 9, no. 6: 1553. https://doi.org/10.3390/cells9061553
APA StyleAlbariqi, M. M. H., Engelsman, S., Eijkelkamp, N., & Höppener, J. W. M. (2020). Amyloid Proteins and Peripheral Neuropathy. Cells, 9(6), 1553. https://doi.org/10.3390/cells9061553