K+ and Ca2+ Channels Regulate Ca2+ Signaling in Chondrocytes: An Illustrated Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
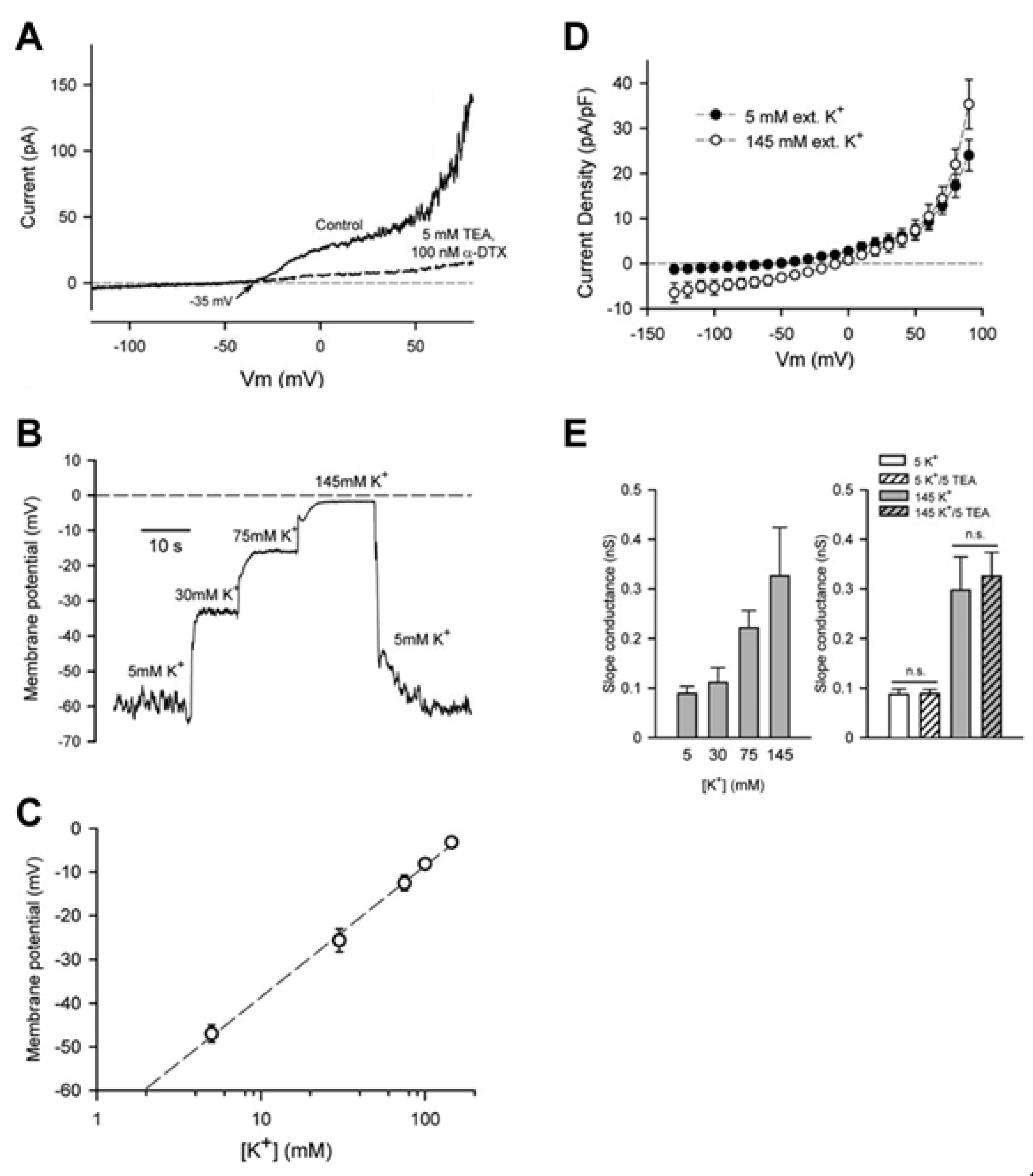
3.1. Chondrocyte Resting Membrane Potential
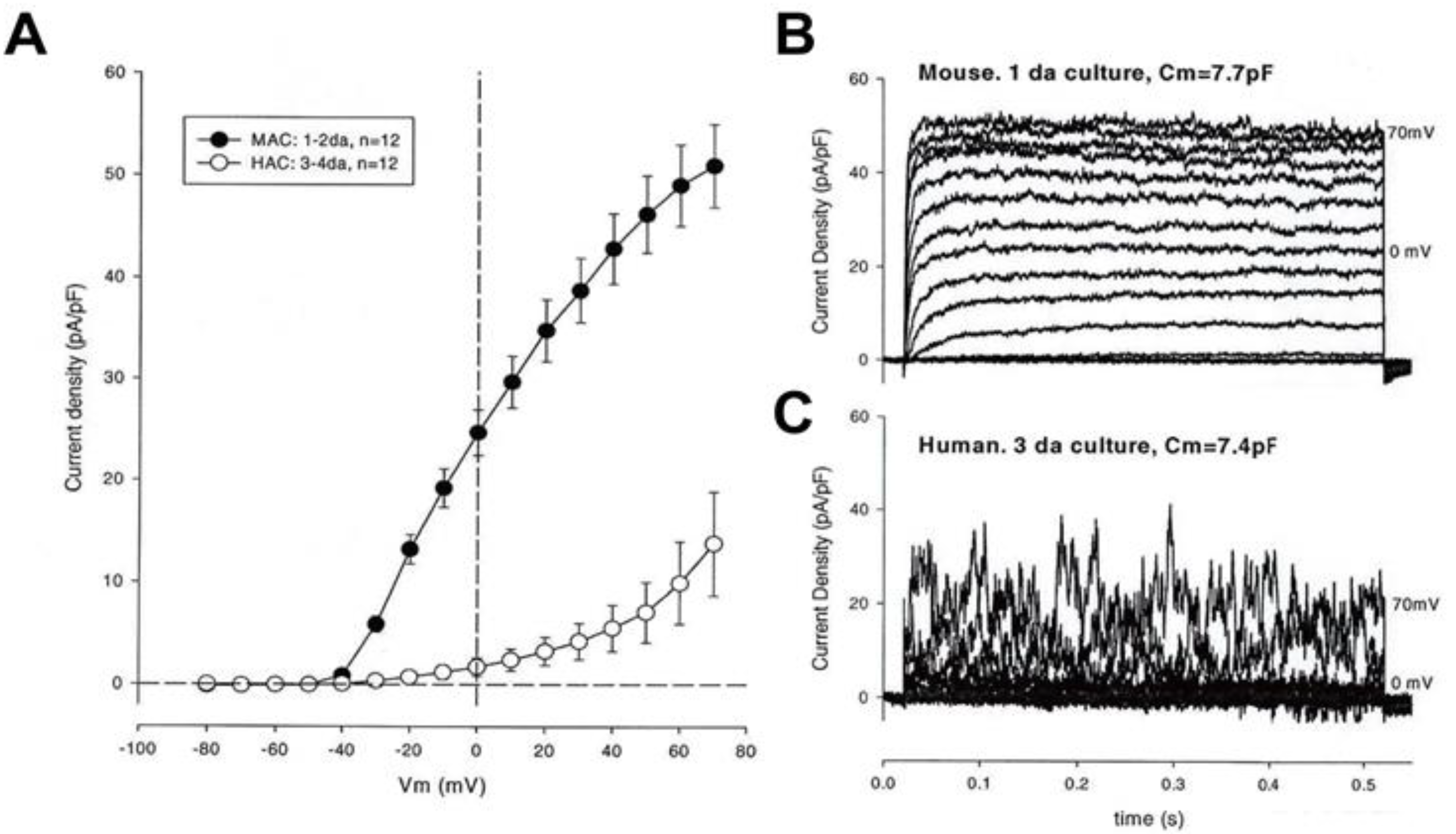
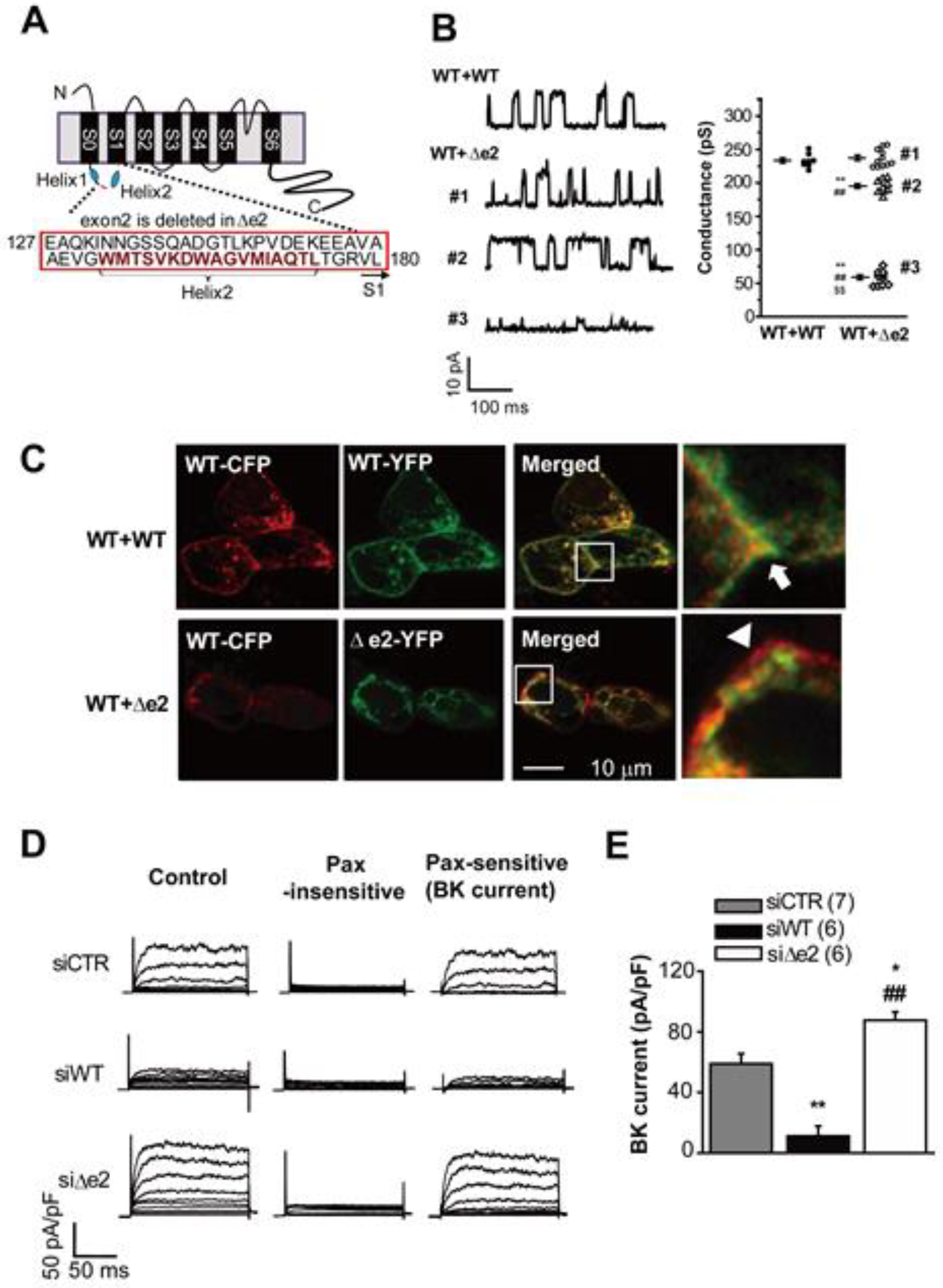
3.2. Large Conductance Ca2+-Activated K+ (BKCa) Channels
3.3. Intermediate Conductance Ca2+-Activated K+ (IKCa or KCa3.1) Channels:
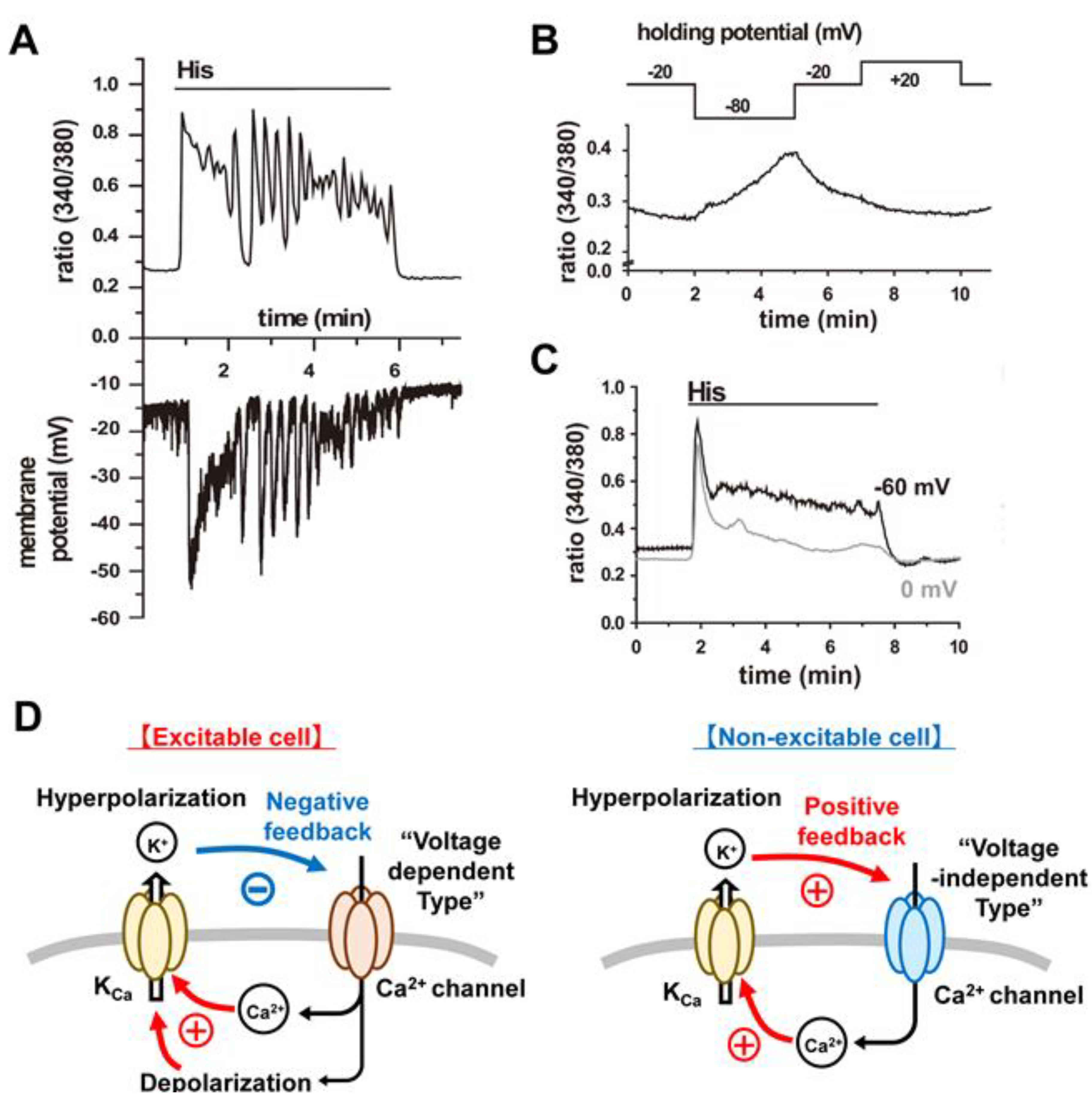
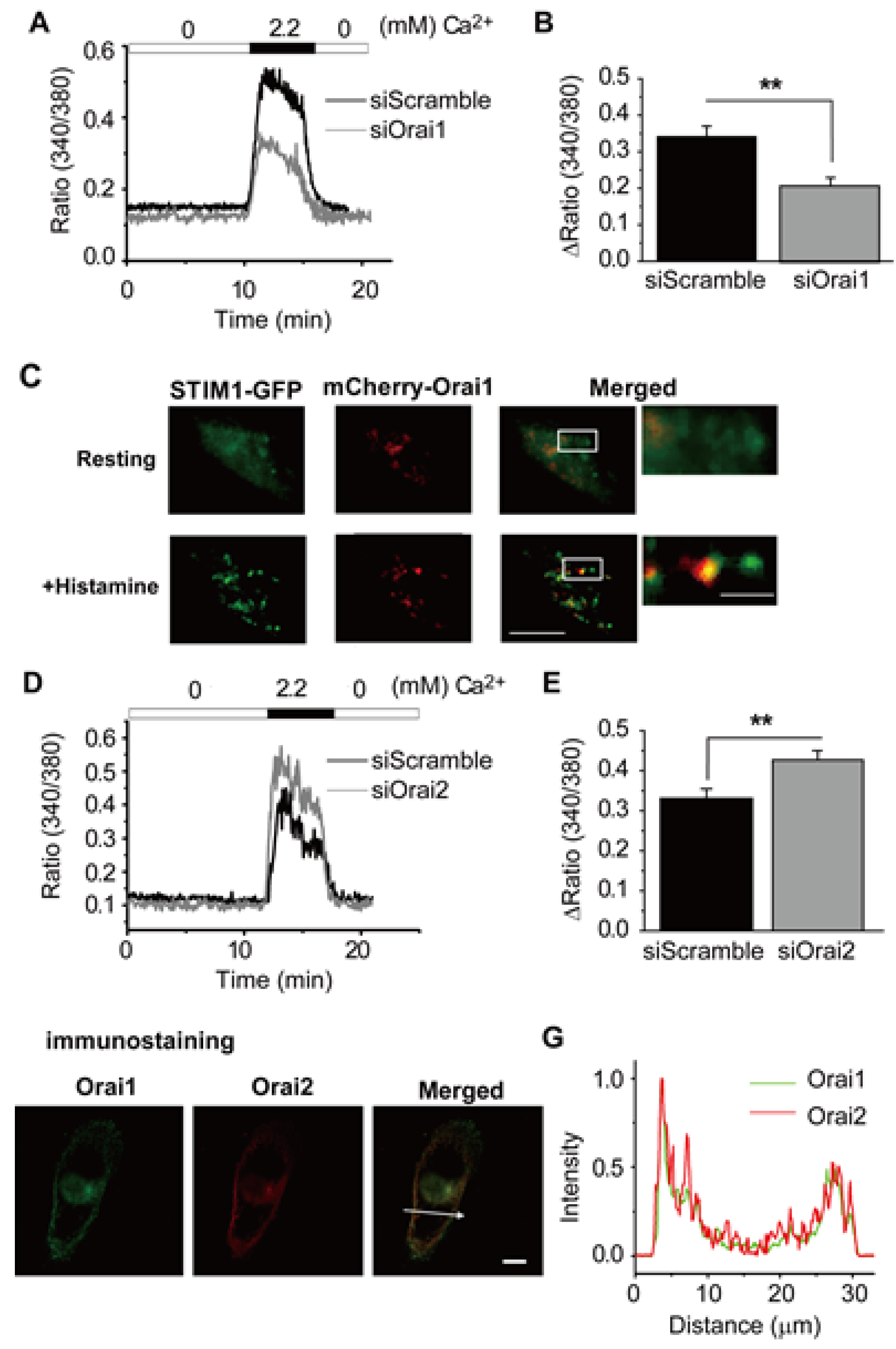
3.4. Ca2+ Release-Activated Ca2+ (CRAC) Channels
- i.
- Can be initiated by a wide variety of signals at the surface membrane (e.g., ATP activation of purinergic receptor subtypes, or stretch) that result in a small net influx of Ca2+ and/or of Ca2+ and Na+.
- ii.
- Almost always requires activation of submembrane phospholipase C as the first step in an intracellular signaling pathway that produces ultimately IP3. IP3 is a potent second messenger that activates significant Ca2+ release from the ER [47].
- iii.
- Is characterized by movement of an ER localized Ca2+ sensor protein (Stromal Interaction Molecule (STIM) 1–2) to discrete junctions of the ER/plasma membrane [48].
- iv.
- Is completed by functional association of these STIM proteins with a second, distinct class of proteins (Orai1–3) at the ER/plasma membrane junctions, resulting in the formation of Ca2+ selective channels [49,50]. These hybrid channels are responsible for the maintained Ca2+ influx and related non-inactivating inward current, denoted ICRAC or ISOCE [50].
- i.
- There is a marked hyperpolarization of plasma membrane due to activation of one or more of the subtypes of KCa channels that are expressed.
- ii.
- The chondrocyte responds to the change in [Ca2+]i, by sometimes generating [Ca2+]i waves or oscillations [51]. These can activate cascades of Ca2+-dependent enzymes, (CaMK2 [6,7], NFAT, and calcineurin [8,11]); enhanced secretion of cytokines, catabolic factors and paracrine substances [52,53]; increased Ca2+-dependent secretion of essential extracellular matrix ([2,10,38,54]) and significant changes in cellular transcription activity [10,54] and/or altered proliferation as well as differentiation [11,51,55].
4. Discussion
4.1. Main Findings
- i.
- Ligand (ATP or histamine) triggered release of Ca2+ from one or more intracellular stores (e.g., ER).
- ii.
- Targeted translocation of a specific intracellular protein (STIM1) from the ER to discrete spatial locations near the surface membrane (the ER-plasma membrane junction).
- iii.
- STIM1-induced conformational changes in a second partner protein (Orai1) and resulting formation of ion channels that readily allow Ca2+ to enter the chondrocyte over quite extended (s) time periods. This is referred to as SOCE, and takes place through CRAC channels.
- iv.
- Alterations (hyperpolarization) in chondrocyte ER triggered by increases in [Ca2+]i and resulting augmentation of Ca2+ influx then initiates or promotes functionally important enzymatic cascades or intracellular regulatory pathways (Ca2+-dependent phosphorylation/dephosphorylation).
4.2. Relationship to Previous Reports of Other Ion Channels and Transporters in Adult Chondrocytes
4.2.1. TRP Channels and Piezo Stretch Sensitive Channels
4.2.2. Ca2+ Channels in Mammalian Chondrocytes
4.2.3. Na+/K+ Pump Expression in Mammalian Chondrocytes
- i.
- ii.
- iii.
- The net outward current generated by Na+/K+ pump turnover, although small (perhaps 10 pA) is capable of hyperpolarizing the membrane potential of the chondrocyte by 10–15 mV, as we have shown using mathematical modeling approaches [21].
4.2.4. Connexin and Pannexin-Based Channels and Signaling
4.2.5. Cl− Channels in Chondrocytes
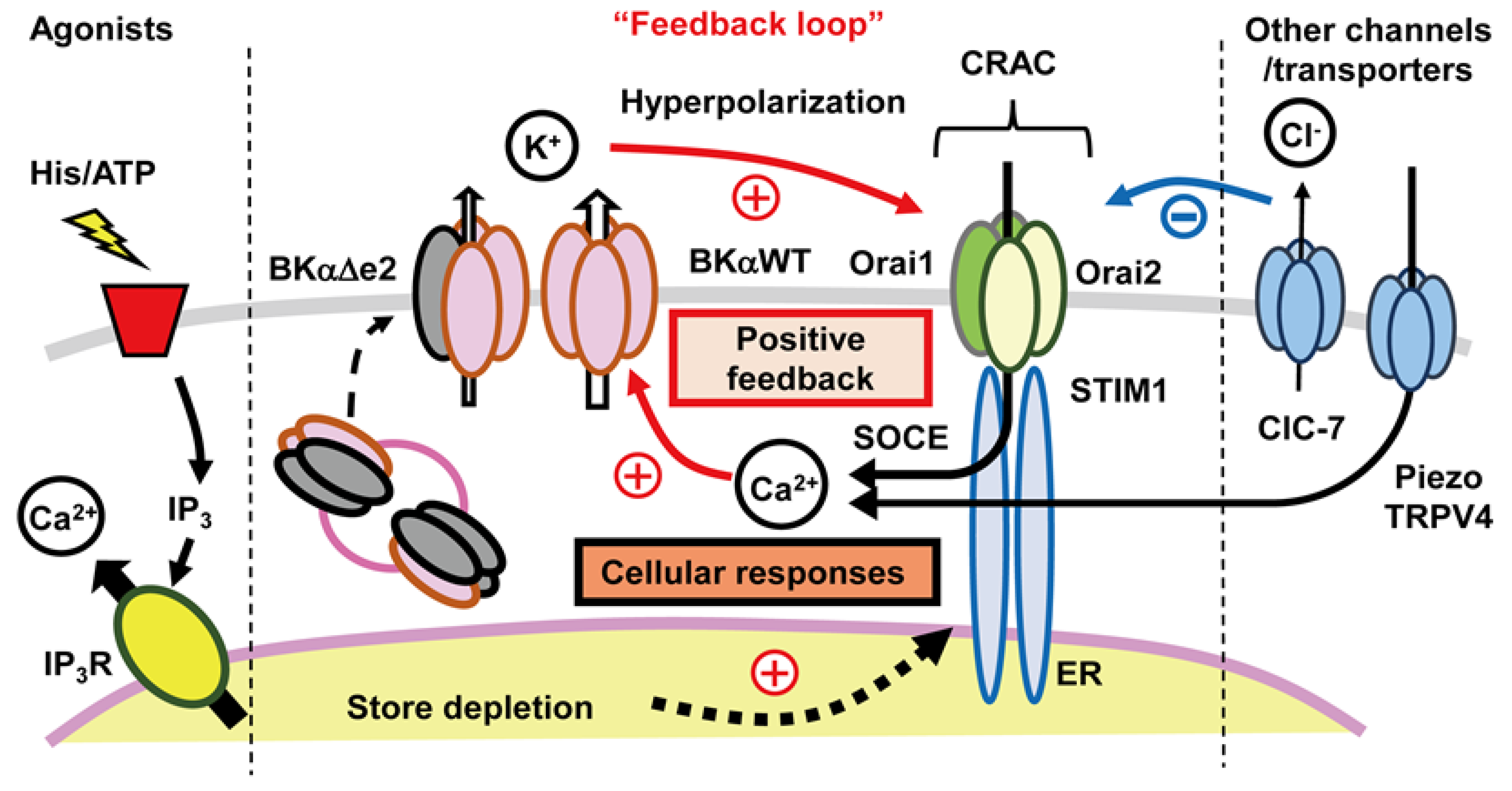
4.3. Functional Coupling between CRAC Channels and KCa Channels:
4.3.1. An Important Feedback Loop
4.3.2. Further Evaluation of Functional Coupling of K+ and Ca2+ Fluxes
- i.
- To use a standard capability of the voltage clamp method in a detailed study of the SOC current, ICRAC, activation and dynamics at fixed membrane potentials within the range that a ligand such as ATP produces when it activates KCa channels.
- ii.
- To evaluate and then implement a novel approach for regulating chondrocyte membrane potential by incorporating optogenetic tools [90], such as synthetic light-sensitive channels including K+ channels [91] into chondrocytes in primary culture. This has the advantages of avoiding disruption of the chondrocyte membrane by patch seal formation and allowing repetitive activation of K+ channels while also assaying changes in [Ca2+]i.
- iii.
- To improve throughput of data acquisition using methods that allow ligand-induced changes in chondrocyte ER in populations of isolated cells. It may be possible to monitor and calibrate a signal obtained during flow cytometry assays [92] to provide absolute or near absolute values of the chondrocyte membrane potential. A number of different synthetic or protein-based voltage-sensitive dyes can be evaluated and considered some of which have quite favorable signal-to-noise ratios [90].
- iv.
- To utilize a Systems Biology approach, incorporating an additional set of measurements and calculations. The results would further evaluate the applicability and validity of key Ca2+-dependent steps in the diagram shown in Figure 6. Insights from ‘semi quantitative assays’ of [Ca2+]i levels at baseline together with parameters describing ligand-induced transients and/or oscillations are much needed. These can be obtained by using recently published analytical software [93]. In other cell and tissue systems, a strong emphasis on details of [Ca2+]i transient waveforms has yielded interesting insights into some aspects of [Ca2+]i homeostasis [94]. This type of platform-based medium throughput analysis can be put in context and new experiments can be designed by combining these approaches with a mathematical model for ligand-based Ca2+ influx, [Ca2+]i release and buffering as well as Ca2+ extrusion. The Hille group [95] have developed and used this type of rationale and mathematical modeling in their studies of Ca2+ homeostasis in the PC-12 cell line.
- v.
- One shortcoming of our working hypothesis, as outlined in Figure 6, is that it does not take full account of the fact that what we denote as distinct ‘ion channels’ almost certainly need to be thought of as ‘ion channel signaling complexes’. This distinction can be illustrated, and interesting new experiments can be planned by re-thinking some key properties of what we have described in this review as the large conductance Ca2+-activated K+ (BKCa) channel and the intermediate conductance Ca2+-activated K+ (IKCa) channel.
4.4. Future Perspectives
4.4.1. Limitation of the Usage of Chondrocyte Cell Line
4.4.2. The Chondron vs. the Chondrocyte
4.4.3. Extracellular Matrix Interaction with Ion Channels
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lewis, R.; Gomez Alvarez, C.B.; Rayman, M.; Lanham-New, S.; Woolf, A.; Mobasheri, A. Strategies for optimising musculoskeletal health in the 21st century. BMC Musculoskelet. Disord. 2019, 20, 164. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.B.; Schmidt, T.A.; Sachse, F.B.; Boyle, D.; Firestein, G.S.; Giles, W.R. Cellular electrophysiological principles that modulate secretion from synovial fibroblasts. J. Physiol. 2017, 595, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobasheri, A.; Batt, M. An update on the pathophysiology of osteoarthritis. Ann. Phys. Rehabil. Med. 2016, 59, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shen, J.; Zhao, W.; Wang, T.; Han, L.; Hamilton, J.L.; Im, H.J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef]
- Sun, M.M.; Beier, F.; Pest, M.A. Recent developments in emerging therapeutic targets of osteoarthritis. Curr. Opin. Rheumatol. 2017, 29, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, S.; Hosaka, Y.; Okada, K.; Mori, D.; Yano, F.; Kobayashi, H.; Taniguchi, Y.; Mori, Y.; Okuma, T.; Chang, S.H.; et al. Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl. Acad. Sci. USA 2015, 112, 3080–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Jin, Z.; Zhang, H.; Piao, S.; Lu, J.; Bai, L. The Transient receptor potential channel, vanilloid 5, induces chondrocyte apoptosis via Ca2+ CaMKII-dependent MAPK and Akt/ mTOR pathways in a rat osteoarthritis model. Cell. Physiol. Biochem. 2018, 51, 2309–2323. [Google Scholar] [CrossRef]
- Yao, W.; Han, Q.; Wang, L.; Niu, Z. Ropivacaine relieves pain and prevents chondrocyte degradation probably through Calcineurin/NFAT1 signaling pathway in osteoarthritis rats. Eur. J. Pharmacol. 2018, 818, 518–524. [Google Scholar] [CrossRef]
- Fu, K.; Robbins, S.R.; McDougall, J.J. Osteoarthritis: The genesis of pain. Rheumatology 2018, 57, iv43–iv50. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Li, G.; Huang, Y.; Fu, Z.; Song, X.; Chen, C.; Yang, L. Synergistically regulated spontaneous calcium signaling is attributed to cartilaginous extracellular matrix metabolism. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Matta, C.; Fodor, J.; Szijgyarto, Z.; Juhasz, T.; Gergely, P.; Csernoch, L.; Zakany, R. Cytosolic free Ca2+ concentration exhibits a characteristic temporal pattern during in vitro cartilage differentiation: A possible regulatory role of calcineurin in Ca-signalling of chondrogenic cells. Cell Calcium 2008, 44, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.; Zhou, Y.; Polson, S.W.; Wan, L.Q.; Wang, M.; Han, L.; Wang, L.; Lu, X.L. Identification of chondrocyte genes and signaling pathways in response to acute joint inflammation. Sci. Rep. 2019, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Castillo, K.; Carrasquel-Ursulaez, W.; Sepulveda, R.V.; Gonzalez-Nilo, F.; Gonzalez, C.; Alvarez, O. Molecular determinants of BK channel functional diversity and functioning. Physiol. Rev. 2017, 97, 39–87. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.B.; Kondo, C.; Belke, D.D.; Giles, W.R. Two-pore domain K+ channels regulate membrane potential of isolated human articular chondrocytes. J. Physiol. 2011, 589, 5071–5089. [Google Scholar] [CrossRef]
- Clark, R.B.; Hatano, N.; Kondo, C.; Belke, D.D.; Brown, B.S.; Kumar, S.; Votta, B.J.; Giles, W.R. Voltage-gated K+ currents in mouse articular chondrocytes regulate membrane potential. Channels (Austin) 2010, 4, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Funabashi, K.; Ohya, S.; Yamamura, H.; Hatano, N.; Muraki, K.; Giles, W.; Imaizumi, Y. Accelerated Ca2+ entry by membrane hyperpolarization due to Ca2+-activated K+ channel activation in response to histamine in chondrocytes. Am. J. Physiol. Cell Physiol. 2010, 298, C786–C797. [Google Scholar] [CrossRef] [Green Version]
- Inayama, M.; Suzuki, Y.; Yamada, S.; Kurita, T.; Yamamura, H.; Ohya, S.; Giles, W.R.; Imaizumi, Y. Orai1-Orai2 complex is involved in store-operated calcium entry in chondrocyte cell lines. Cell Calcium 2015, 57, 337–347. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ohya, S.; Yamamura, H.; Giles, W.R.; Imaizumi, Y. A new splice variant of large conductance Ca2+-activated K+ (BK) channel α subunit alters human chondrocyte function. J. Biol. Chem. 2016, 291, 24247–24260. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.R.; Duncan, N.A.; Giles, W.R.; Clark, R.B. A voltage-dependent K+ current contributes to membrane potential of acutely isolated canine articular chondrocytes. J. Physiol. 2004, 557, 93–104. [Google Scholar] [CrossRef]
- Maleckar, M.M.; Clark, R.B.; Votta, B.; Giles, W.R. The resting potential and K+ currents in primary human articular chondrocytes. Front. Physiol. 2018, 9, 974. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; May, H.; Mobasheri, A.; Barrett-Jolley, R. Chondrocyte channel transcriptomics: Do microarray data fit with expression and functional data? Channels (Austin) 2013, 7, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.; Feetham, C.H.; Barrett-Jolley, R. Cell volume regulation in chondrocytes. Cell. Physiol. Biochem. 2011, 28, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Barrett-Jolley, R.; Lewis, R.; Fallman, R.; Mobasheri, A. The emerging chondrocyte channelome. Front. Physiol. 2010, 1, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, H.; Suzuki, Y.; Imaizumi, Y. Physiological and pathological functions of Cl− channels in chondrocytes. Biol. Pharm. Bull. 2018, 41, 1145–1151. [Google Scholar] [CrossRef]
- Lewis, R.; Asplin, K.E.; Bruce, G.; Dart, C.; Mobasheri, A.; Barrett-Jolley, R. The role of the membrane potential in chondrocyte volume regulation. J. Cell. Physiol. 2011, 226, 2979–2986. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Kolski-Andreaco, A.; Sankaranarayanan, A.; Sabatier, J.M.; Shakkottai, V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem. 2007, 14, 1437–1457. [Google Scholar] [CrossRef]
- Varga, Z.; Juhasz, T.; Matta, C.; Fodor, J.; Katona, E.; Bartok, A.; Olah, T.; Sebe, A.; Csernoch, L.; Panyi, G.; et al. Switch of voltage-gated K+ channel expression in the plasma membrane of chondrogenic cells affects cytosolic Ca2+-oscillations and cartilage formation. PLoS ONE 2011, 6, e27957. [Google Scholar] [CrossRef] [Green Version]
- Zweifach, A.; Lewis, R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA. 1993, 90, 6295–6299. [Google Scholar] [CrossRef] [Green Version]
- Church, P.J.; Stanley, E.F. Single L-type calcium channel conductance with physiological levels of calcium in chick ciliary ganglion neurons. J. Physiol. 1996, 496 Pt 1, 59–68. [Google Scholar] [CrossRef]
- Singh, A.K.; McMillan, J.; Bukiya, A.N.; Burton, B.; Parrill, A.L.; Dopico, A.M. Multiple cholesterol recognition/interaction amino acid consensus (CRAC) motifs in cytosolic C tail of Slo1 subunit determine cholesterol sensitivity of Ca2+- and voltage-gated K+ (BK) channels. J. Biol. Chem. 2012, 287, 20509–20521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshi, T.; Tian, Y.; Xu, R.; Heinemann, S.H.; Hou, S. Mechanism of the modulation of BK potassium channel complexes with different auxiliary subunit compositions by the omega-3 fatty acid DHA. Proc. Natl. Acad. Sci. USA 2013, 110, 4822–4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.X. Lipid-dependent gating of ion channels. In Protein-Lipid Interactions: Perspectives, Techniques and Challenges; Catala, A., Ed.; Nova Science Publishers: New York, NY, USA, 2018; p. 196. [Google Scholar]
- Ding, J.; Li, J.; Xue, C.; Wu, K.; Ouyang, W.; Zhang, D.; Yan, Y.; Huang, C. Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in Beas-2B cells. J. Biol. Chem. 2006, 281, 24405–24413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.C.; Chai, C.Y.; Chen, W.C.; Hou, M.F.; Wang, Y.S.; Chiu, Y.C.; Lu, S.R.; Chang, W.C.; Juo, S.H.; Wang, J.Y. Histamine regulates cyclooxygenase 2 gene activation through Orai1-mediated NFκB activation in lung cancer cells. Cell Calcium 2011, 50, 27–35. [Google Scholar] [CrossRef]
- Morimoto, T.; Ohya, S.; Hayashi, H.; Onozaki, K.; Imaizumi, Y. Cell-cycle-dependent regulation of Ca2+-activated K+ channel in Jurkat T-lymphocyte. J. Pharmacol. Sci. 2007, 104, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Ghanshani, S.; Wulff, H.; Miller, M.J.; Rohm, H.; Neben, A.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 2000, 275, 37137–37149. [Google Scholar] [CrossRef] [Green Version]
- Kondo, C.; Clark, R.B.; Al-Jezani, N.; Kim, T.Y.; Belke, D.; Banderali, U.; Szerencsei, R.T.; Jalloul, A.H.; Schnetkamp, P.P.M.; Spitzer, K.W.; et al. ATP triggers a robust intracellular [Ca2+ ]-mediated signalling pathway in human synovial fibroblasts. Exp. Physiol. 2018, 103, 1101–1122. [Google Scholar] [CrossRef]
- Millward-Sadler, S.J.; Wright, M.O.; Flatman, P.W.; Salter, D.M. ATP in the mechanotransduction pathway of normal human chondrocytes. Biorheology 2004, 41, 567–575. [Google Scholar]
- Ostrom, R.S.; Gregorian, C.; Insel, P.A. Cellular release of and response to ATP as key determinants of the set-point of signal transduction pathways. J. Biol. Chem. 2000, 275, 11735–11739. [Google Scholar] [CrossRef] [Green Version]
- Kumahashi, N.; Naitou, K.; Nishi, H.; Oae, K.; Watanabe, Y.; Kuwata, S.; Ochi, M.; Ikeda, M.; Uchio, Y. Correlation of changes in pain intensity with synovial fluid adenosine triphosphate levels after treatment of patients with osteoarthritis of the knee with high-molecular-weight hyaluronic acid. Knee 2011, 18, 160–164. [Google Scholar] [CrossRef]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xiang, M. Targeting potassium channels Kv1.3 and KCa3.1: Routes to selective immunomodulators in autoimmune disorder treatment? Pharmacotherapy 2013, 33, 515–528. [Google Scholar] [CrossRef]
- Gallo, E.M.; Cante-Barrett, K.; Crabtree, G.R. Lymphocyte calcium signaling from membrane to nucleus. Nat. Immunol. 2006, 7, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Mocsar, G.; Sebestyen, V.; Volko, J.; Papp, F.; Toth, K.; Damjanovich, S.; Panyi, G.; Waldmann, T.A.; Bodnar, A.; et al. Membrane Potential Distinctly Modulates Mobility and Signaling of IL-2 and IL-15 Receptors in T Cells. Biophys. J. 2018, 114, 2473–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, D.O.; Foskett, J.K. Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: A single-channel point of view. Cell Calcium 2015, 58, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahalan, M.D. STIMulating store-operated Ca2+ entry. Nat. Cell Biol. 2009, 11, 669–677. [Google Scholar] [CrossRef]
- Qiu, R.; Lewis, R.S. Structural features of STIM and Orai underlying store-operated calcium entry. Curr. Opin. Cell Biol. 2019, 57, 90–98. [Google Scholar] [CrossRef]
- Amcheslavsky, A.; Wood, M.L.; Yeromin, A.V.; Parker, I.; Freites, J.A.; Tobias, D.J.; Cahalan, M.D. Molecular biophysics of Orai store-operated Ca2+ channels. Biophys. J. 2015, 108, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Fodor, J.; Matta, C.; Olah, T.; Juhasz, T.; Takacs, R.; Toth, A.; Dienes, B.; Csernoch, L.; Zakany, R. Store-operated calcium entry and calcium influx via voltage-operated calcium channels regulate intracellular calcium oscillations in chondrogenic cells. Cell Calcium 2013, 54, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.A.; Park, B.H.; Yoon, H.J.; Lee, J.Y.; Song, J.H.; Kim, H.A.; Cho, C.S.; Kim, W.U. Calcineurin modulates the catabolic and anabolic activity of chondrocytes and participates in the progression of experimental osteoarthritis. Arthritis Rheum. 2007, 56, 2299–2311. [Google Scholar] [CrossRef]
- Little, C.B.; Hughes, C.E.; Curtis, C.L.; Jones, S.A.; Caterson, B.; Flannery, C.R. Cyclosporin A inhibition of aggrecanase-mediated proteoglycan catabolism in articular cartilage. Arthritis Rheum. 2002, 46, 124–129. [Google Scholar] [CrossRef]
- O’Conor, C.J.; Leddy, H.A.; Benefield, H.C.; Liedtke, W.B.; Guilak, F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc. Natl. Acad. Sci. USA 2014, 111, E1316–E1321. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, S.; Wakabayashi, M.; Ohno, T.; Amano, K.; Ooishi, R.; Sugahara, T.; Shiojiri, S.; Tashiro, K.; Suzuki, Y.; Nishimura, R.; et al. Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J. Biol. Chem. 2007, 282, 32158–32167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, M.N.; Leddy, H.A.; Votta, B.J.; Kumar, S.; Levy, D.S.; Lipshutz, D.B.; Lee, S.H.; Liedtke, W.; Guilak, F. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum. 2009, 60, 3028–3037. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.L.; Votta, B.J.; Kumar, S.; Liedtke, W.; Guilak, F. Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: Age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum. 2010, 62, 2973–2983. [Google Scholar] [CrossRef]
- Servin-Vences, M.R.; Moroni, M.; Lewin, G.R.; Poole, K. Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. Elife 2017, 6, e21074. [Google Scholar] [CrossRef]
- Lee, W.; Guilak, F.; Liedtke, W. Role of Piezo Channels in Joint Health and Injury. Curr. Top. Membr. 2017, 79, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Du, G.; Li, L.; Zhang, X.; Liu, J.; Hao, J.; Zhu, J.; Wu, H.; Chen, W.; Zhang, Q. Roles of TRPV4 and piezo channels in stretch-evoked Ca2+ response in chondrocytes. Exp. Biol. Med. (Maywoodn.J.) 2020, 245, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coste, B.; Xiao, B.; Santos, J.S.; Syeda, R.; Grandl, J.; Spencer, K.S.; Kim, S.E.; Schmidt, M.; Mathur, J.; Dubin, A.E.; et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 2012, 483, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Leddy, H.A.; Chen, Y.; Lee, S.H.; Zelenski, N.A.; McNulty, A.L.; Wu, J.; Beicker, K.N.; Coles, J.; Zauscher, S.; et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E5114–E5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.K.; Wouters, W.; Clark, A.; Herzog, W. Mechanically induced calcium signaling in chondrocytes in situ. J. Orthop. Res. 2012, 30, 475–481. [Google Scholar] [CrossRef]
- Parekh, A.B.; Muallem, S. Ca2+ signalling and gene regulation. Cell Calcium 2011, 49, 279. [Google Scholar] [CrossRef]
- Wu, Q.Q.; Chen, Q. Mechanoregulation of chondrocyte proliferation, maturation, and hypertrophy: Ion-channel dependent transduction of matrix deformation signals. Exp. Cell Res. 2000, 256, 383–391. [Google Scholar] [CrossRef]
- Atsuta, Y.; Tomizawa, R.R.; Levin, M.; Tabin, C.J. L-type voltage-gated Ca2+ channel CaV1.2 regulates chondrogenesis during limb development. Proc. Natl. Acad. Sci. USA 2019, 116, 21592–21601. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, P.P.; Parajuli, A.; Price, C.; Wang, L.; Duncan, R.L.; Kirn-Safran, C.B. Inhibition of T-Type voltage sensitive calcium channel reduces load-induced OA in mice and suppresses the catabolic effect of bone mechanical stress on chondrocytes. PLoS ONE 2015, 10, e0127290. [Google Scholar] [CrossRef]
- Mobasheri, A.; Errington, R.J.; Golding, S.; Hall, A.C.; Urban, J.P. Characterization of the Na+, K+-ATPase in isolated bovine articular chondrocytes; molecular evidence for multiple alpha and beta isoforms. Cell Biol. Int. 1997, 21, 201–212. [Google Scholar] [CrossRef]
- Mobasheri, A. Correlation between [Na+], [glycosaminoglycan] and Na+/K+ pump density in the extracellular matrix of bovine articular cartilage. Physiol. Res. 1998, 47, 47–52. [Google Scholar]
- Shakibaei, M.; Mobasheri, A. Beta1-integrins co-localize with Na, K-ATPase, epithelial sodium channels (ENaC) and voltage activated calcium channels (VACC) in mechanoreceptor complexes of mouse limb-bud chondrocytes. Histol. Histopathol. 2003, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proc. Natl. Acad. Sci. USA 2003, 100, 6257–6262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova, V.V.; Petrov, A.M.; Matchkov, V.V.; Bouzinova, E.V.; Vasiliev, A.N.; Benziane, B.; Zefirov, A.L.; Chibalin, A.V.; Heiny, J.A.; Krivoi, I.I. Distinct alpha2 Na, K-ATPase membrane pools are differently involved in early skeletal muscle remodeling during disuse. J. Gen. Physiol. 2016, 147, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, M.M.; McGlashan, S.R.; Garcia, M.; Jensen, C.G.; Poole, C.A. Articular chondrocytes express connexin 43 hemichannels and P2 receptors-a putative mechanoreceptor complex involving the primary cilium? J. Anat. 2009, 214, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Knight, M.M. Cyclic loading opens hemichannels to release ATP as part of a chondrocyte mechanotransduction pathway. J. Orthop. Res. 2010, 28, 510–515. [Google Scholar] [CrossRef]
- Matta, C.; Fodor, J.; Miosge, N.; Takacs, R.; Juhasz, T.; Rybaltovszki, H.; Toth, A.; Csernoch, L.; Zakany, R. Purinergic signalling is required for calcium oscillations in migratory chondrogenic progenitor cells. Pflug. Arch 2015, 467, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Bond, S.R.; Lau, A.; Penuela, S.; Sampaio, A.V.; Underhill, T.M.; Laird, D.W.; Naus, C.C. Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J. Bone Miner. Res. 2011, 26, 2911–2922. [Google Scholar] [CrossRef]
- Iwamoto, T.; Nakamura, T.; Doyle, A.; Ishikawa, M.; de Vega, S.; Fukumoto, S.; Yamada, Y. Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation. J. Biol. Chem. 2010, 285, 18948–18958. [Google Scholar] [CrossRef] [Green Version]
- Funabashi, K.; Fujii, M.; Yamamura, H.; Ohya, S.; Imaizumi, Y. Contribution of chloride channel conductance to the regulation of resting membrane potential in chondrocytes. J. Pharmacol. Sci. 2010, 113, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, T.; Yamamura, H.; Suzuki, Y.; Giles, W.R.; Imaizumi, Y. The ClC-7 chloride channel is downregulated by hypoosmotic stress in human chondrocytes. Mol. Pharm. 2015, 88, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, K.; Toyoda, F.; Staunton, C.A.; Maeda, T.; Okumura, N.; Matsuura, H.; Matsusue, Y.; Imai, S.; Barrett-Jolley, R. Activation of a chondrocyte volume-sensitive Cl− conductance prior to macroscopic cartilage lesion formation in the rabbit knee anterior cruciate ligament transection osteoarthritis model. Osteoarthr. Cartil. 2016, 24, 1786–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, N.; Imai, S.; Toyoda, F.; Isoya, E.; Kumagai, K.; Matsuura, H.; Matsusue, Y. Regulatory role of tyrosine phosphorylation in the swelling-activated chloride current in isolated rabbit articular chondrocytes. J. Physiol. 2009, 587, 3761–3776. [Google Scholar] [CrossRef] [PubMed]
- Isoya, E.; Toyoda, F.; Imai, S.; Okumura, N.; Kumagai, K.; Omatsu-Kanbe, M.; Kubo, M.; Matsuura, H.; Matsusue, Y. Swelling-activated Cl− current in isolated rabbit articular chondrocytes: Inhibition by arachidonic acid. J. Pharmacol. Sci. 2009, 109, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Bertram, K.L.; Krawetz, R.J. Osmolarity regulates chondrogenic differentiation potential of synovial fluid derived mesenchymal progenitor cells. Biochem. Biophys. Res. Commun. 2012, 422, 455–461. [Google Scholar] [CrossRef]
- Leanza, L.; Biasutto, L.; Manago, A.; Gulbins, E.; Zoratti, M.; Szabo, I. Intracellular ion channels and cancer. Front. Physiol. 2013, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Yadav, G.P.; Zheng, H.; Yang, Q.; Douma, L.G.; Bloom, L.B.; Jiang, Q.X. Secretory granule protein chromogranin B (CHGB) forms an anion channel in membranes. Life Sci. Alliance 2018, 1, e201800139. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.E.; Venkatachalam, V. Bringing bioelectricity to light. Annu. Rev. Biophys. 2014, 43, 211–232. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, C.; Alberio, L.; Gazzarrini, S.; Aquila, M.; Romano, E.; Cermenati, S.; Zuccolini, P.; Petersen, J.; Beltrame, M.; Van Etten, J.L.; et al. Optogenetics. Engineering of a light-gated potassium channel. Science 2015, 348, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Klapperstuck, T.; Glanz, D.; Klapperstuck, M.; Wohlrab, J. Methodological aspects of measuring absolute values of membrane potential in human cells by flow cytometry. Cytometry Part A 2009, 75, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.; Mikolajewicz, N.; Komarova, S.V.; Khadra, A. Systematic Characterization of Dynamic Parameters of Intracellular Calcium Signals. Front. Physiol. 2016, 7, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, C.M.; Yang, J.H.; Santolini, M.; Lusis, A.J.; Weiss, J.N.; Karma, A. The Ca2+ transient as a feedback sensor controlling cardiomyocyte ionic conductances in mouse populations. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Duman, J.G.; Chen, L.; Hille, B. Calcium transport mechanisms of PC12 cells. J. Gen. Physiol. 2008, 131, 307–323. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Heinemann, S.H.; Hoshi, T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiol. (Bethesdamd.) 2009, 24, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Zahan, O.M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Bautista, L.; Castro, M.J.; Lopez-Barneo, J.; Castellano, A. Hypoxia inducible factor-2alpha stabilization and maxi-K+ channel beta1-subunit gene repression by hypoxia in cardiac myocytes: Role in preconditioning. Circ. Res. 2009, 104, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.T.; Kim, Y.M.; Adams, E.; Lyu, S.C.; Alvira, C.M.; Cornfield, D.N. Hypoxia-inducible factor-1alpha regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L352–L359. [Google Scholar] [CrossRef] [Green Version]
- Pfander, D.; Gelse, K. Hypoxia and osteoarthritis: How chondrocytes survive hypoxic environments. Curr. Opin. Rheumatol. 2007, 19, 457–462. [Google Scholar] [CrossRef]
- Whitt, J.P.; McNally, B.A.; Meredith, A.L. Differential contribution of Ca2+ sources to day and night BK current activation in the circadian clock. J. Gen. Physiol. 2018, 150, 259–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Aldrich, R.W. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature 2010, 466, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Suzuki, Y.; Yamamura, H.; Giles, W.R.; Imaizumi, Y. Roles of LRRC26 as an auxiliary gamma1-subunit of large-conductance Ca2+-activated K+ channels in bronchial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019. [Google Scholar] [CrossRef]
- Cheong, A.; Bingham, A.J.; Li, J.; Kumar, B.; Sukumar, P.; Munsch, C.; Buckley, N.J.; Neylon, C.B.; Porter, K.E.; Beech, D.J.; et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol. Cell 2005, 20, 45–52. [Google Scholar] [CrossRef]
- Coleman, N.; Brown, B.M.; Olivan-Viguera, A.; Singh, V.; Olmstead, M.M.; Valero, M.S.; Kohler, R.; Wulff, H. New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1. Mol. Pharm. 2014, 86, 342–357. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Begenisich, T. Membrane-delimited inhibition of maxi-K channel activity by the intermediate conductance Ca2+-activated K channel. J. Gen. Physiol. 2006, 127, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Kunisada, T.; Miyazaki, M.; Mihara, K.; Gao, C.; Kawai, A.; Inoue, H.; Namba, M. A new human chondrosarcoma cell line (OUMS-27) that maintains chondrocytic differentiation. Int J. Cancer 1998, 77, 854–859. [Google Scholar] [CrossRef]
- Santoro, A.; Conde, J.; Scotece, M.; Abella, V.; Lopez, V.; Pino, J.; Gomez, R.; Gomez-Reino, J.J.; Gualillo, O. Choosing the right chondrocyte cell line: Focus on nitric oxide. J. Orthop. Res. 2015, 33, 1784–1788. [Google Scholar] [CrossRef]
- Finger, F.; Schorle, C.; Zien, A.; Gebhard, P.; Goldring, M.B.; Aigner, T. Molecular phenotyping of human chondrocyte cell lines T/C-28a2, T/C-28a4, and C-28/I2. Arthritis Rheum. 2003, 48, 3395–3403. [Google Scholar] [CrossRef]
- Zhang, Z. Chondrons and the pericellular matrix of chondrocytes. Tissue Eng. Part B Rev. 2015, 21, 267–277. [Google Scholar] [CrossRef]
- Lee, G.M.; Poole, C.A.; Kelley, S.S.; Chang, J.; Caterson, B. Isolated chondrons: A viable alternative for studies of chondrocyte metabolism in vitro. Osteoarthr. Cartil. 1997, 5, 261–274. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.T.; Chang, P.; Granqvist, A.; Boehm, H.; Kramer, A.; Scrimgeour, J.; Curtis, J.E. Spatial organization and mechanical properties of the pericellular matrix on chondrocytes. Biophys. J. 2013, 104, 986–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, R.E.; Sanchez-Adams, J.; Guilak, F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 2014, 39, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Zelenski, N.A.; Leddy, H.A.; Sanchez-Adams, J.; Zhang, J.; Bonaldo, P.; Liedtke, W.; Guilak, F. Type VI collagen regulates pericellular matrix properties, chondrocyte swelling, and mechanotransduction in mouse articular cartilage. Arthritis Rheumatol. 2015, 67, 1286–1294. [Google Scholar] [CrossRef] [Green Version]
- Guilak, F.; Alexopoulos, L.G.; Upton, M.L.; Youn, I.; Choi, J.B.; Cao, L.; Setton, L.A.; Haider, M.A. The pericellular matrix as a transducer of biomechanical and biochemical signals in articular cartilage. Ann. N. Y. Acad. Sci. 2006, 1068, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef] [PubMed]
- DiDomenico, C.D.; Lintz, M.; Bonassar, L.J. Molecular transport in articular cartilage-what have we learned from the past 50 years? Nat. Rev. Rheumatol. 2018, 14, 393–403. [Google Scholar] [CrossRef]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shortt, C.; Zhang, F.; Bater, M.Q.; Cowman, M.K.; Kirsch, T. Extracellular vesicles released from articular chondrocytes play a major role in cell-cell communication. J. Orthop. Res. 2020, 38, 731–739. [Google Scholar] [CrossRef]
- Rellmann, Y.; Dreier, R. Different forms of ER stress in chondrocytes result in short stature disorders and degenerative cartilage diseases: New insights by cartilage-specific ERp57 knockout mice. Oxidative Med. Cell. Longev. 2018, 2018, 8421394. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, Y.; Yamamura, H.; Imaizumi, Y.; Clark, R.B.; Giles, W.R. K+ and Ca2+ Channels Regulate Ca2+ Signaling in Chondrocytes: An Illustrated Review. Cells 2020, 9, 1577. https://doi.org/10.3390/cells9071577
Suzuki Y, Yamamura H, Imaizumi Y, Clark RB, Giles WR. K+ and Ca2+ Channels Regulate Ca2+ Signaling in Chondrocytes: An Illustrated Review. Cells. 2020; 9(7):1577. https://doi.org/10.3390/cells9071577
Chicago/Turabian StyleSuzuki, Yoshiaki, Hisao Yamamura, Yuji Imaizumi, Robert B. Clark, and Wayne R. Giles. 2020. "K+ and Ca2+ Channels Regulate Ca2+ Signaling in Chondrocytes: An Illustrated Review" Cells 9, no. 7: 1577. https://doi.org/10.3390/cells9071577
APA StyleSuzuki, Y., Yamamura, H., Imaizumi, Y., Clark, R. B., & Giles, W. R. (2020). K+ and Ca2+ Channels Regulate Ca2+ Signaling in Chondrocytes: An Illustrated Review. Cells, 9(7), 1577. https://doi.org/10.3390/cells9071577