MHC Class I-Restricted TCR-Transgenic CD4+ T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo

 , , ,
, , ,  , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Isolation of PBMC, CD4+ and CD8+ T Cell Populations
2.3. Retroviral Transduction of T Cell Subsets, Purification, Expansion, and Culture Methods
2.4. FACS Staining and Analysis
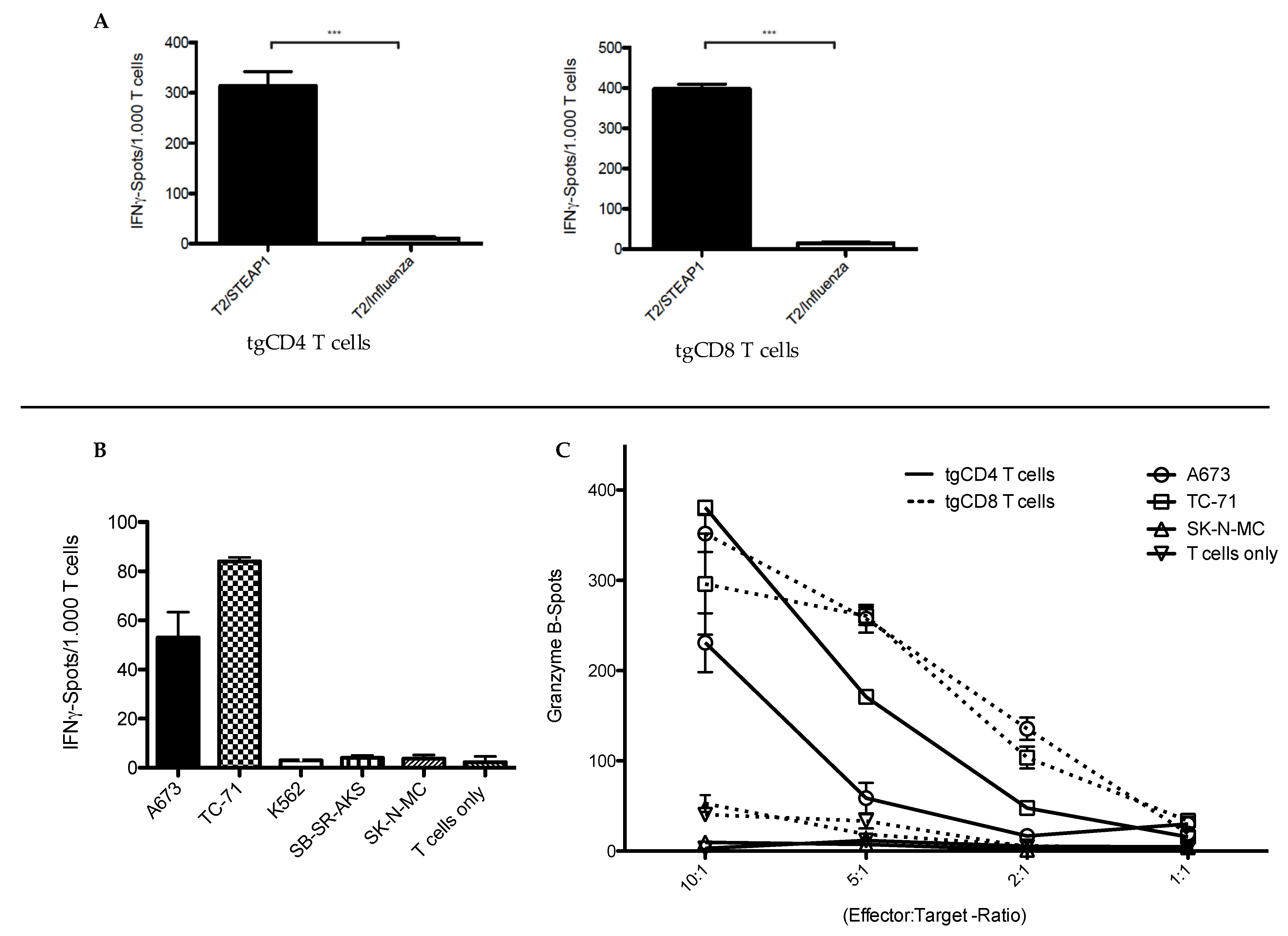
2.5. Functional Characterization of STEAP1130/HLA-A*02:01-Specific TCR Transgenic T Cell Subsets
2.6. Animal Model
2.7. In Vivo Experiments
2.7.1. Local Tumor Control
2.7.2. Experimental Metastasis
2.7.3. Bioluminescence Monitoring of Implanted Tumors
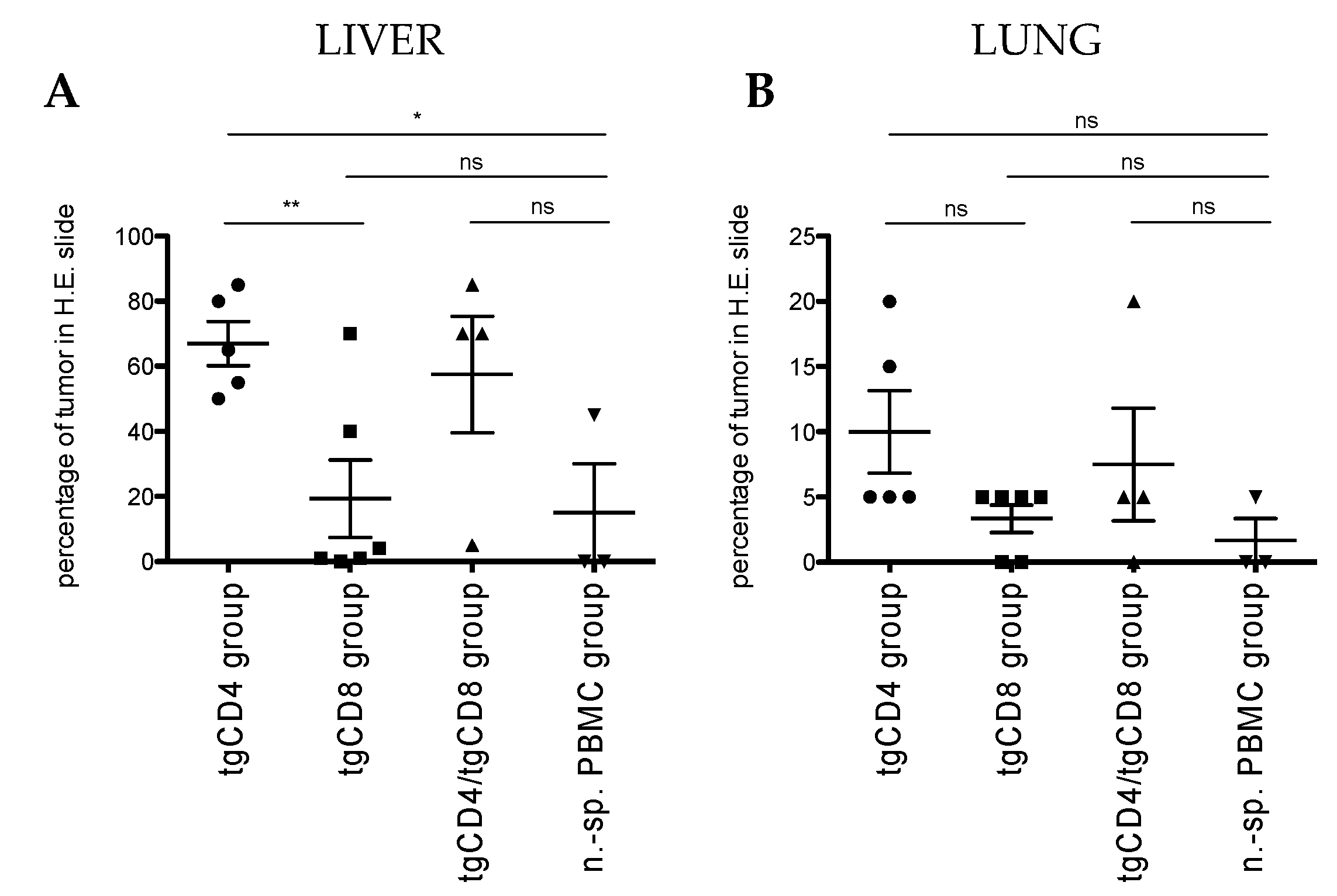
2.8. Histopathological Analysis
2.9. Statistical Analysis
2.10. Analysis of Public Patient Data
3. Results
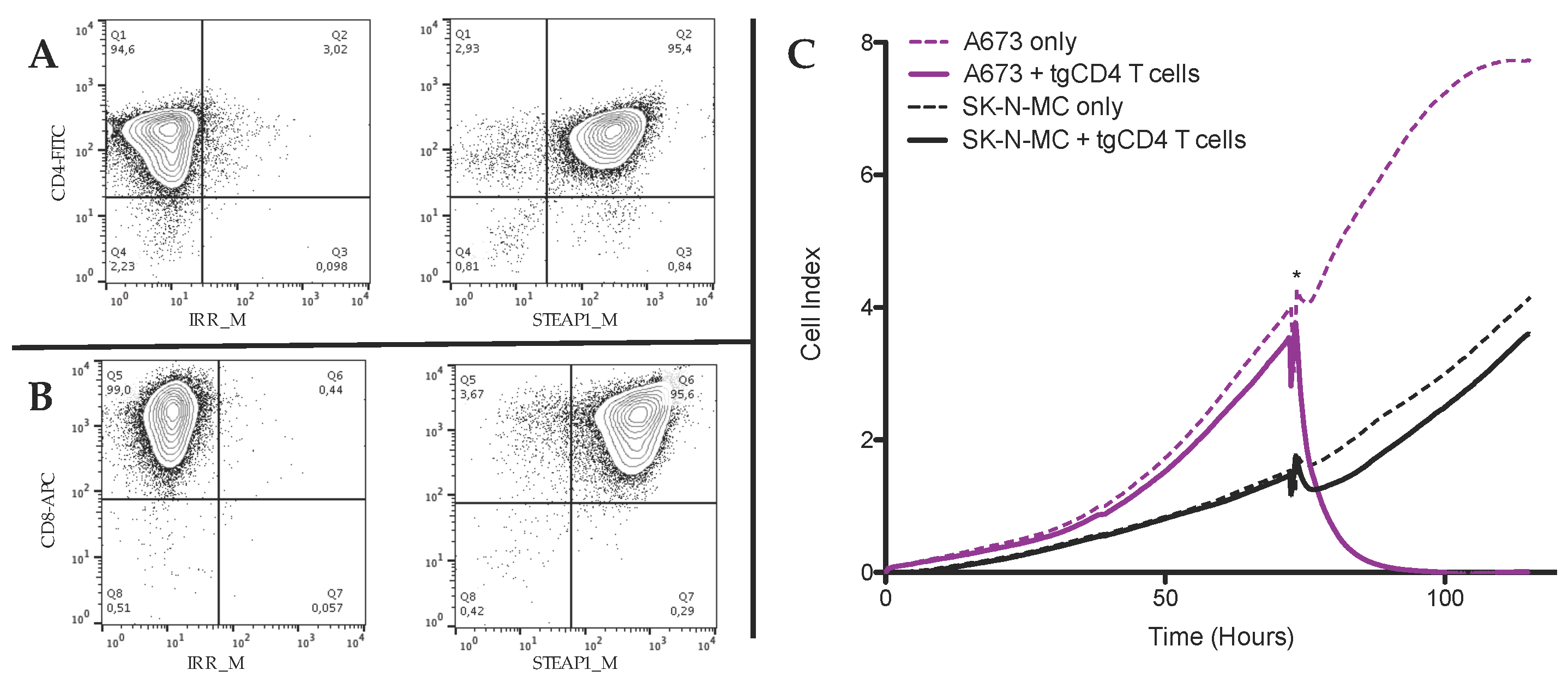
3.1. In Vitro Antitumor Activity of tgCD4+ T Cells Increases Over Time
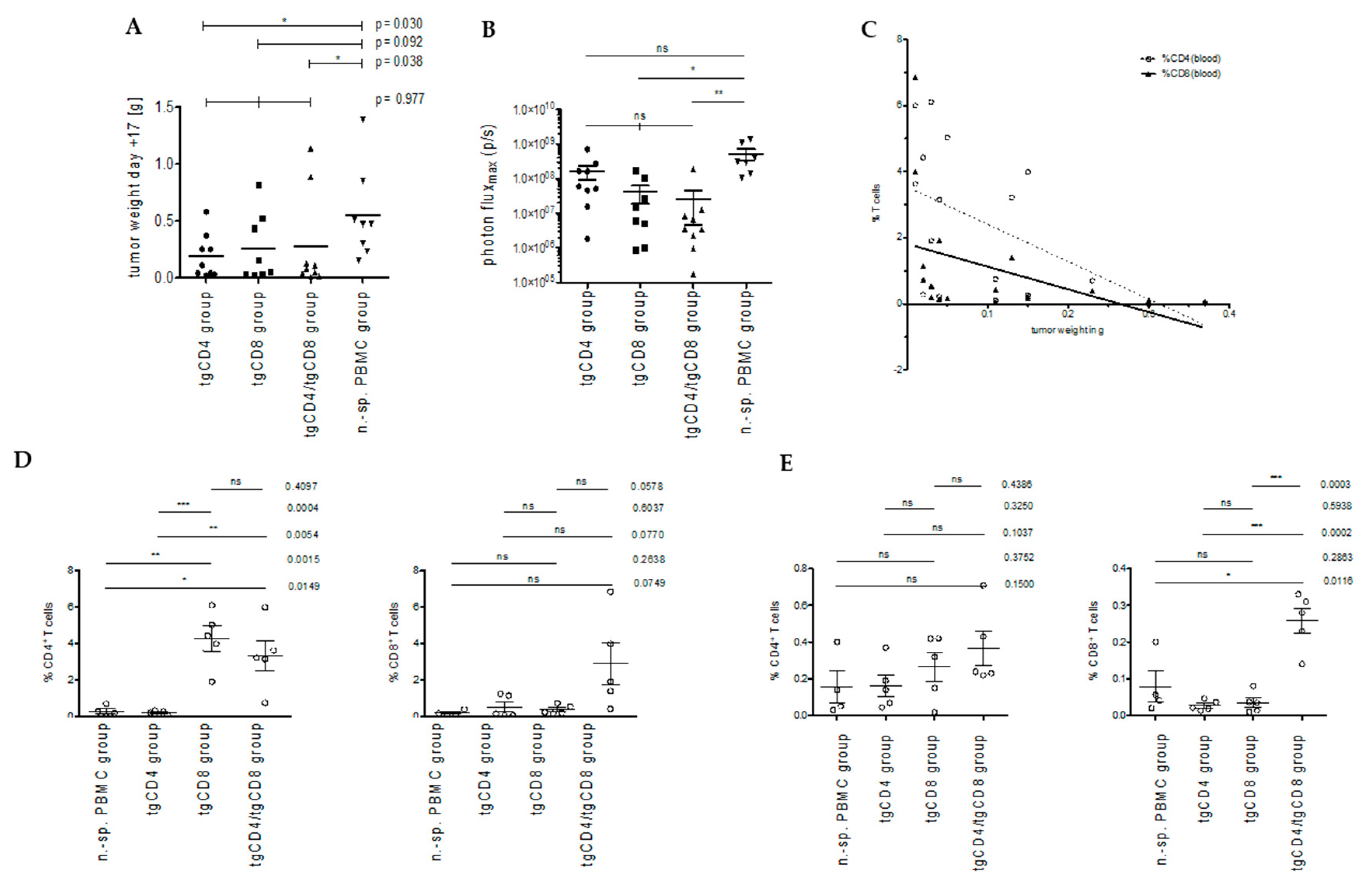
3.2. STEAP1130/HLA-A*02:01-Specific TCR Transgenic CD4+ T Cells Control Tumor Growth in a Xenografted Local Tumor Model
3.2.1. Bioluminescence-Based Analysis of Tumor Burden in Local Tumor Model
3.2.2. Correlation of T Cell Frequencies in Blood and Spleen with Tumor Weight
3.3. STEAP1130/HLA-A*02:01-Specific TCR Transgenic CD4+ T Cells Do Not Control Metastatic Outgrowth in a Model of Experimental Metastasis
3.4. Exploratory Bioinformatic Analysis Reveals a Correlation of CXCL10 and Survival and Points to Differences of T Cell Subset-Specific Transcripts Depending on Primary, Metastatic or Relapsed Site
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Spurny, C.; Kailayangiri, S.; Altvater, B.; Jamitzky, S.; Hartmann, W.; Wardelmann, E.; Ranft, A.; Dirksen, U.; Amler, S.; Hardes, J.; et al. T cell infiltration into Ewing sarcomas is associated with local expression of immune-inhibitory HLA-G. Oncotarget 2018, 9, 6536–6549. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Fukushi, J.; Yamamoto, S.; Matsumoto, Y.; Setsu, N.; Oda, Y.; Yamada, H.; Okada, S.; Watari, K.; Ono, M.; et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am. J. Pathol. 2011, 179, 1157–1170. [Google Scholar] [CrossRef]
- Evdokimova, V.; Ruzanov, P.; Gassmann, H.; Zaidi, S.H.; Peltekova, V.; Heisler, L.E.; McPherson, J.D.; Orlic-Milacic, M.; Specht, K.; Steiger, K.; et al. Exosomes transmit retroelement RNAs to drive inflammation and immunosuppression in Ewing Sarcoma. bioRxiv 2019, 806851. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Schmidt, D.; Harms, D.; Burdach, S. Malignant peripheral neuroectodermal tumours of childhood and adolescence. Virchows Arch. A Pathol. Anat. Histopathol. 1985, 406, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Thiel, U.; Wawer, A.; von Luettichau, I.; Bender, H.U.; Blaeschke, F.; Grünewald, T.G.; Steinborn, M.; Roper, B.; Bonig, H.; Klingebiel, T.; et al. Bone marrow involvement identifies a subgroup of advanced Ewing sarcoma patients with fatal outcome irrespective of therapy in contrast to curable patients with multiple bone metastases but unaffected marrow. Oncotarget 2016, 7, 70959–70968. [Google Scholar] [CrossRef]
- Ferrari, S.; Luksch, R.; Hall, K.S.; Fagioli, F.; Prete, A.; Tamburini, A.; Tienghi, A.; DiGirolamo, S.; Paioli, A.; Abate, M.E.; et al. Post-relapse survival in patients with Ewing sarcoma. Pediatr. Blood Cancer 2015, 62, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Burdach, S.; van Kaick, B.; Laws, H.J.; Ahrens, S.; Haase, R.; Korholz, D.; Pape, H.; Dunst, J.; Kahn, T.; Willers, R.; et al. Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-Cell Transplant Programs at Dusseldorf University Medical Center, Germany and St. Anna Kinderspital, Vienna, Austria. Ann. Oncol. 2000, 11, 1451–1462. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res. 2004, 64, 8213–8221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Muller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 2012, 10, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, T.G.; Bach, H.; Cossarizza, A.; Matsumoto, I. The STEAP protein family: Versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol. Cell 2012, 104, 641–657. [Google Scholar] [CrossRef]
- Schirmer, D.; Grünewald, T.G.; Klar, R.; Schmidt, O.; Wohlleber, D.; Rubio, R.A.; Uckert, W.; Thiel, U.; Bohne, F.; Busch, D.H.; et al. Transgenic antigen-specific, HLA-A*02:01-allo-restricted cytotoxic T cells recognize tumor-associated target antigen STEAP1 with high specificity. Oncoimmunology 2016, 5, e1175795. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, D.; de Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; van Eggermond, M.C.; van den Elsen, P.J.; Taminiau, A.H.; Ottaviano, L.; Schaefer, K.L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231. [Google Scholar] [CrossRef]
- Berghuis, D.; Santos, S.J.; Baelde, H.J.; Taminiau, A.H.; Egeler, R.M.; Schilham, M.W.; Hogendoorn, P.C.; Lankester, A.C. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J. Pathol. 2011, 223, 347–357. [Google Scholar] [CrossRef]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Stahl, D.; Gentles, A.J.; Thiele, R.; Gutgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology 2019, 8, e1674113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrends, T.; Spanjaard, A.; Pilzecker, B.; Babala, N.; Bovens, A.; Xiao, Y.; Jacobs, H.; Borst, J. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017, 47, 848–861.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhang, H.; Shi, X.; Xiao, X.; Fan, Y.; Minze, L.J.; Wang, J.; Ghobrial, R.M.; Xia, J.; Sciammas, R.; et al. Ablation of Transcription Factor IRF4 Promotes Transplant Acceptance by Driving Allogenic CD4(+) T Cell Dysfunction. Immunity 2017, 47, 1114–1128.e6. [Google Scholar] [CrossRef] [PubMed]
- Schober, S.J.; Thiede, M.; Schirmer, D.; Wohlleber, D.; Richter, G.; Busch, D.H.; Thiel, U.; Burdach, S.E. Abstract LB-106: Allorepertoire-derived HLA class I/peptide-specific T cell receptor transgenic CD4+ T cells mediate antitumor responses in Ewing sarcoma mimicking allo-rejection. Cancer Res. 2018, 78, LB-106. [Google Scholar] [CrossRef]
- Hay, K.A.; Turtle, C.J. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs 2017, 77, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, L.H.; Goodall, J.C.; Gaston, J.S. Major histocompatibility complex class I-restricted alloreactive CD4+ T cells. Immunology 2004, 112, 54–63. [Google Scholar] [CrossRef]
- Soto, C.M.; Stone, J.D.; Chervin, A.S.; Engels, B.; Schreiber, H.; Roy, E.J.; Kranz, D.M. MHC-class I-restricted CD4 T cells: A nanomolar affinity TCR has improved anti-tumor efficacy in vivo compared to the micromolar wild-type TCR. Cancer Immunol. Immunother. 2013, 62, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.A.; Gao, L.; Ahmadi, M.; Ghorashian, S.; Barros, R.D.; Pospori, C.; Holler, A.; Wright, G.; Thomas, S.; Topp, M.; et al. Human MHC Class I-restricted high avidity CD4(+) T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology 2013, 2, e22590. [Google Scholar] [CrossRef] [Green Version]
- Blaeschke, F.; Thiel, U.; Kirschner, A.; Thiede, M.; Rubio, R.A.; Schirmer, D.; Kirchner, T.; Richter, G.H.S.; Mall, S.; Klar, R.; et al. Human HLA-A*02:01/CHM1+ allo-restricted T cell receptor transgenic CD8+ T cells specifically inhibit Ewing sarcoma growth in vitro and in vivo. Oncotarget 2016, 7, 43267–43280. [Google Scholar] [CrossRef]
- Busch, D.H.; Pilip, I.M.; Vijh, S.; Pamer, E.G. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity 1998, 8, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, D. Generation of Transgenic, Antigen-Specific, Allogeneic, HLA-A*02:01-Restricted, Cytotoxic T Cells Directed Against Ewing Sarcoma Specific Target Antigen STEAP1. Technical University of Munich, University Library TUM. Available online: https://mediatum.ub.tum.de/doc/1286472/1286472.pdf (accessed on 29 June 2020).
- Kirschner, A.; Thiede, M.; Grünewald, T.G.; Alba Rubio, R.; Richter, G.H.; Kirchner, T.; Busch, D.H.; Burdach, S.; Thiel, U. Pappalysin-1 T cell receptor transgenic allo-restricted T cells kill Ewing sarcoma in vitro and in vivo. Oncoimmunology 2017, 6, e1273301. [Google Scholar] [CrossRef] [Green Version]
- R Core Team (2019). R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- R2: Genomics Analysis and Visualization Platform.Academic Medical Center Amsterdam. Available online: http://r2.amc.nl (accessed on 24 April 2020).
- Peper, J.K.; Schuster, H.; Loffler, M.W.; Schmid-Horch, B.; Rammensee, H.G.; Stevanovic, S. An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J. Immunol. Methods 2014, 405, 192–198. [Google Scholar] [CrossRef]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, J.; Tsuji, T.; Luescher, I.F.; Shiku, H.; Mineno, J.; Okamoto, S.; Old, L.J.; Shrikant, P.; Gnjatic, S.; Odunsi, K. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci. Rep. 2015, 5, 14896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorduijn, E.M.; Sluijter, M.; Salvatori, D.C.; Silvestri, S.; Maas, S.; Arens, R.; Ossendorp, F.; van der Burg, S.H.; van Hall, T. CD4(+) T Cell and NK Cell Interplay Key to Regression of MHC Class I(low) Tumors upon TLR7/8 Agonist Therapy. Cancer Immunol. Res. 2017, 5, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Martinez, F.; Loza, M.I.; Dominguez, E. Beyond Tumor Suppression: Senescence in Cancer Stemness and Tumor Dormancy. Cells 2020, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Muller-Hermelink, N.; Braumuller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Franco-Luzon, L.; Garcia-Mulero, S.; Sanz-Pamplona, R.; Melen, G.; Ruano, D.; Lassaletta, A.; Madero, L.; Gonzalez-Murillo, A.; Ramirez, M. Genetic and Immune Changes Associated with Disease Progression under the Pressure of Oncolytic Therapy in A Neuroblastoma Outlier Patient. Cancers 2020, 12, 1104. [Google Scholar] [CrossRef]
- Wightman, S.C.; Uppal, A.; Pitroda, S.P.; Ganai, S.; Burnette, B.; Stack, M.; Oshima, G.; Khan, S.; Huang, X.; Posner, M.C.; et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br. J. Cancer 2015, 113, 327–335. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schober, S.J.; Thiede, M.; Gassmann, H.; Prexler, C.; Xue, B.; Schirmer, D.; Wohlleber, D.; Stein, S.; Grünewald, T.G.P.; Busch, D.H.; et al. MHC Class I-Restricted TCR-Transgenic CD4+ T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo. Cells 2020, 9, 1581. https://doi.org/10.3390/cells9071581
Schober SJ, Thiede M, Gassmann H, Prexler C, Xue B, Schirmer D, Wohlleber D, Stein S, Grünewald TGP, Busch DH, et al. MHC Class I-Restricted TCR-Transgenic CD4+ T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo. Cells. 2020; 9(7):1581. https://doi.org/10.3390/cells9071581
Chicago/Turabian StyleSchober, Sebastian J., Melanie Thiede, Hendrik Gassmann, Carolin Prexler, Busheng Xue, David Schirmer, Dirk Wohlleber, Stefanie Stein, Thomas G. P. Grünewald, Dirk H. Busch, and et al. 2020. "MHC Class I-Restricted TCR-Transgenic CD4+ T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo" Cells 9, no. 7: 1581. https://doi.org/10.3390/cells9071581
APA StyleSchober, S. J., Thiede, M., Gassmann, H., Prexler, C., Xue, B., Schirmer, D., Wohlleber, D., Stein, S., Grünewald, T. G. P., Busch, D. H., Richter, G. H. S., Burdach, S. E. G., & Thiel, U. (2020). MHC Class I-Restricted TCR-Transgenic CD4+ T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo. Cells, 9(7), 1581. https://doi.org/10.3390/cells9071581