The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
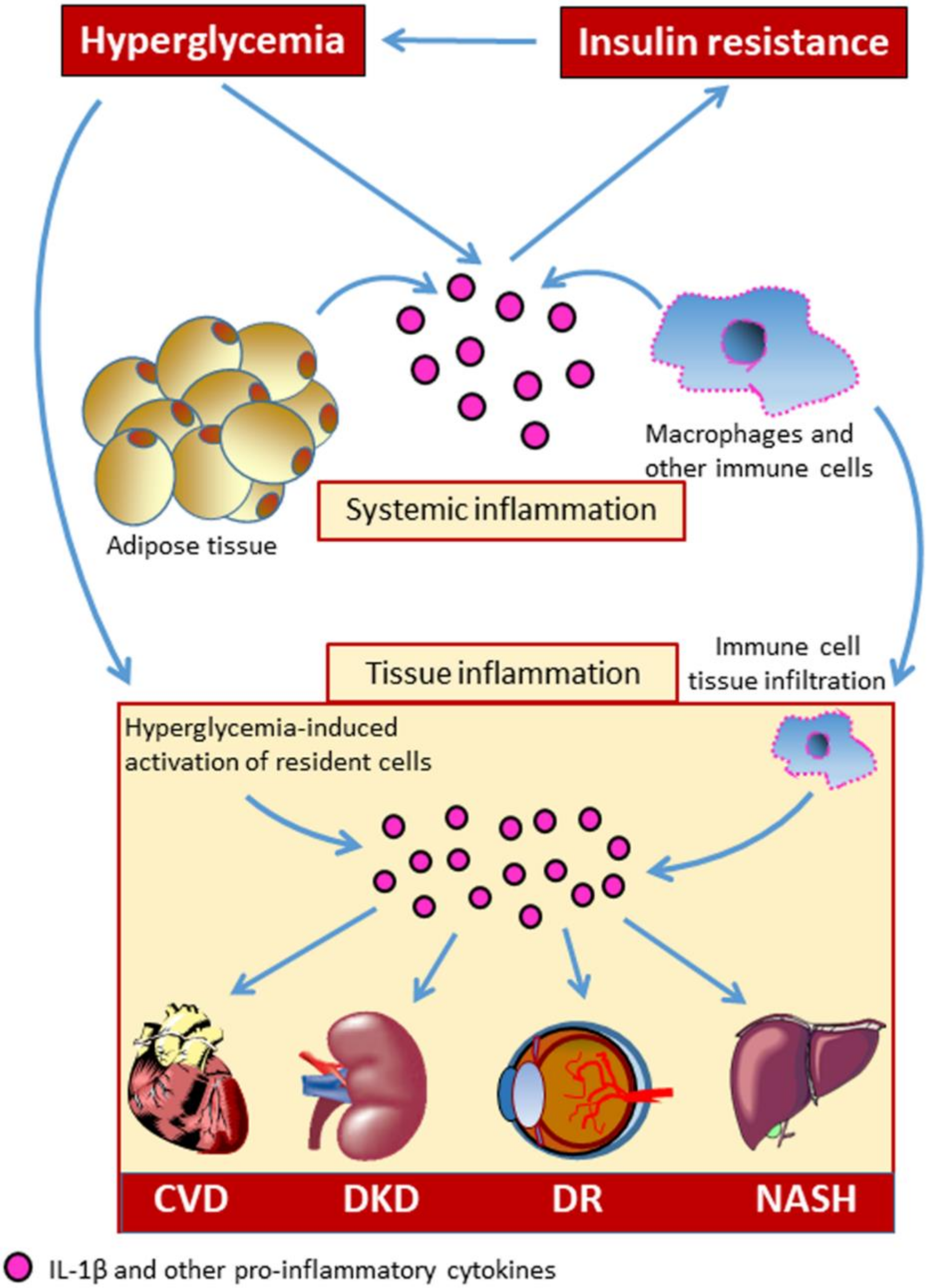
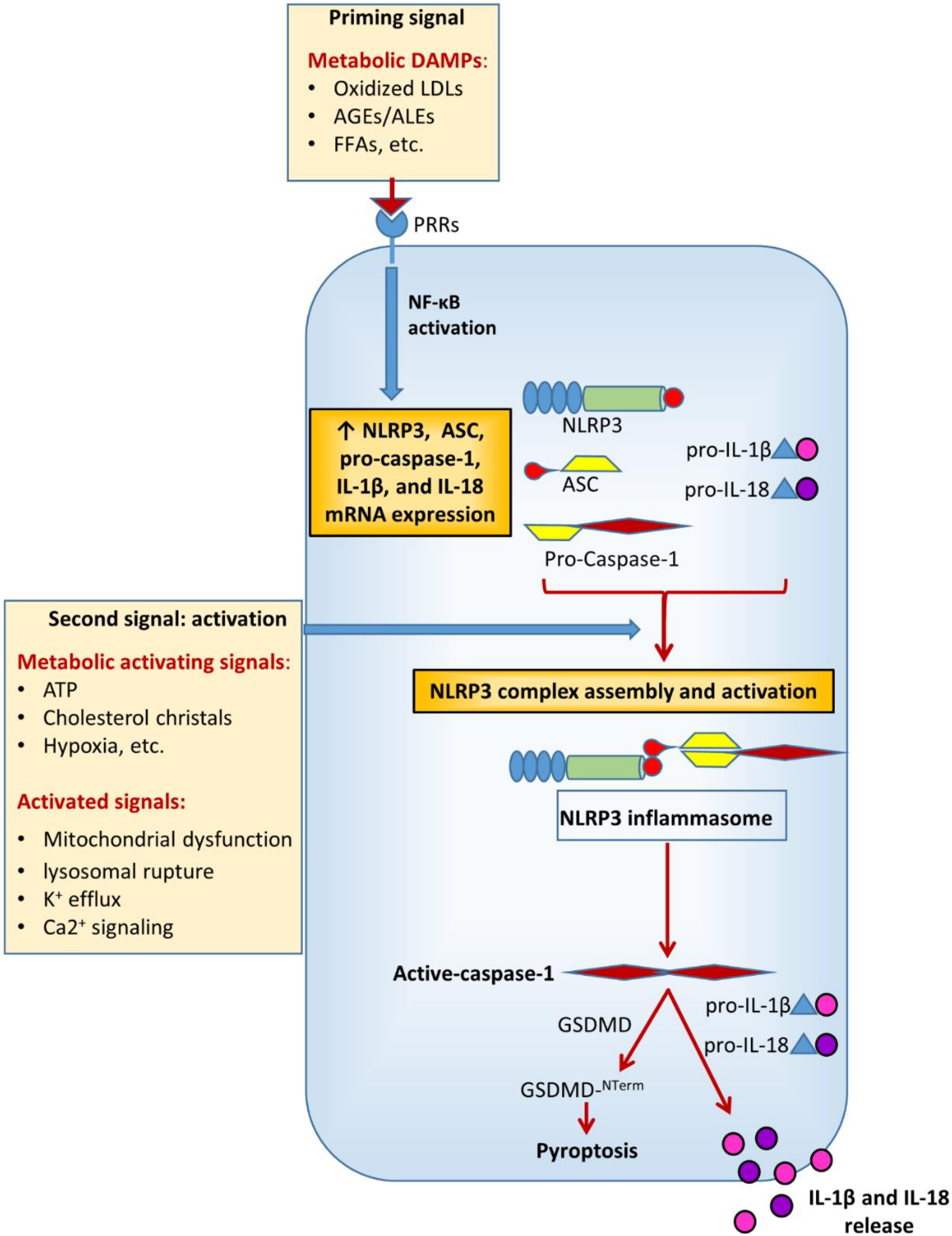
2. Activation and Regulation of the Inflammasome in Diabetes Mellitus and Related Metabolic Disorders
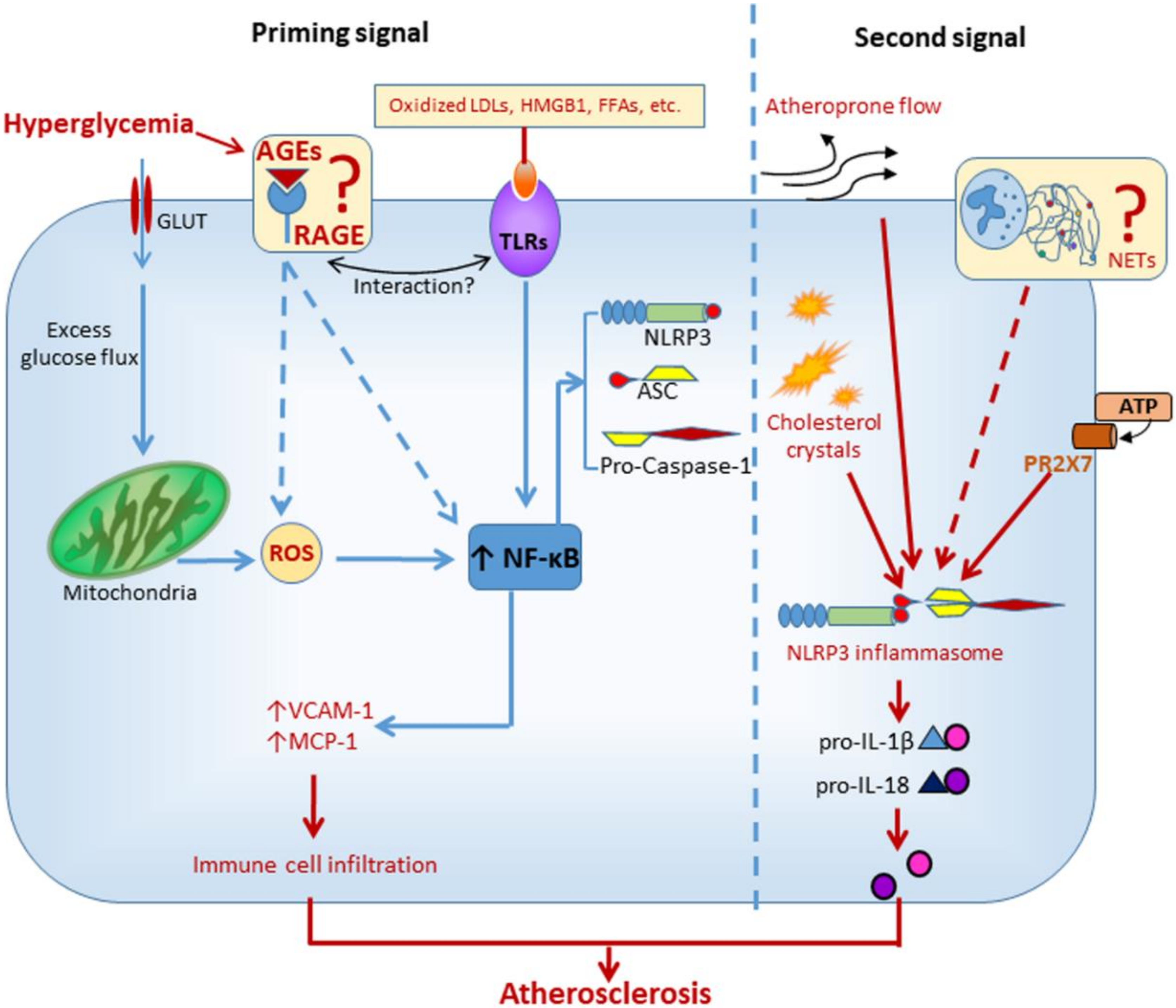
3. Diabetic Cardiovascular Disease
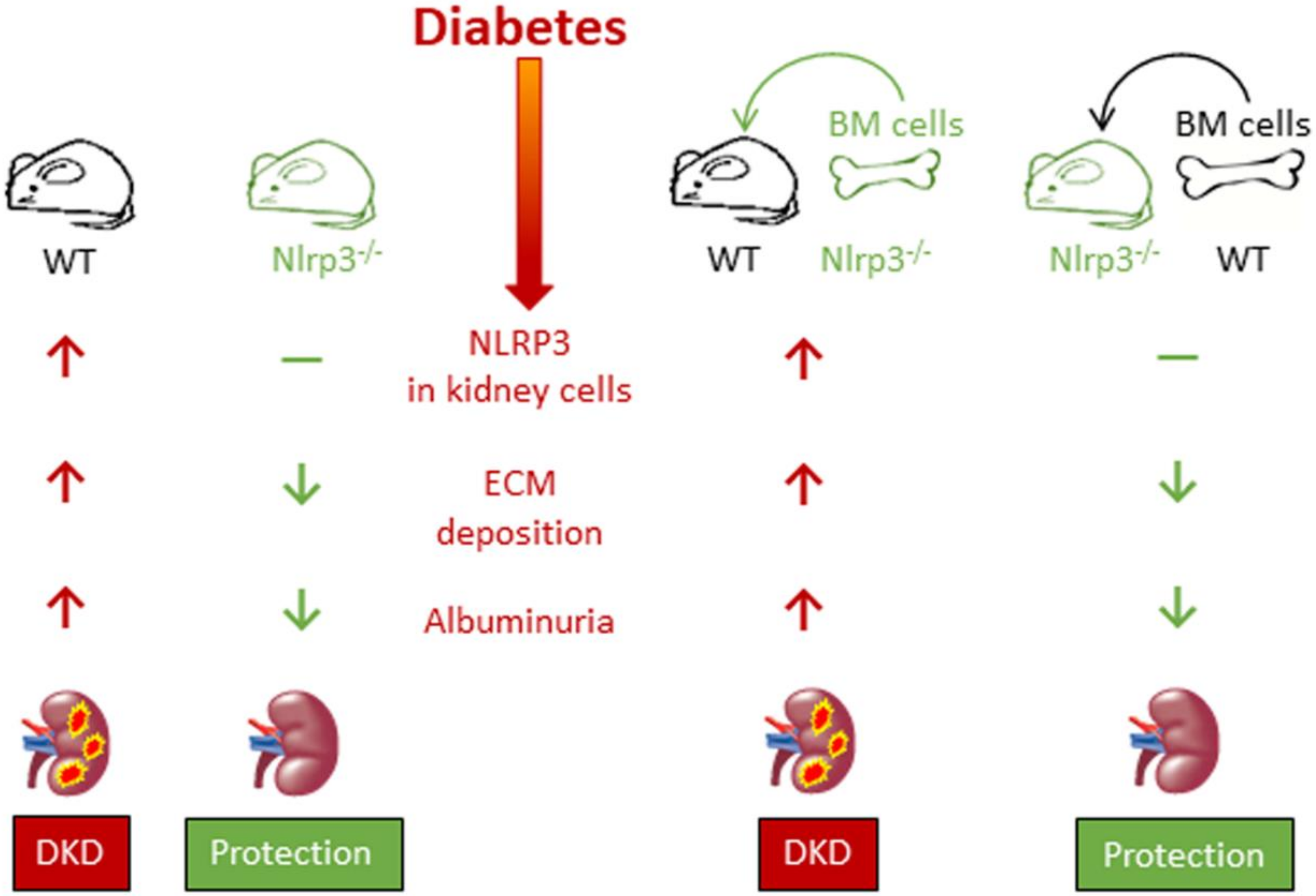
4. Diabetic Kidney Disease
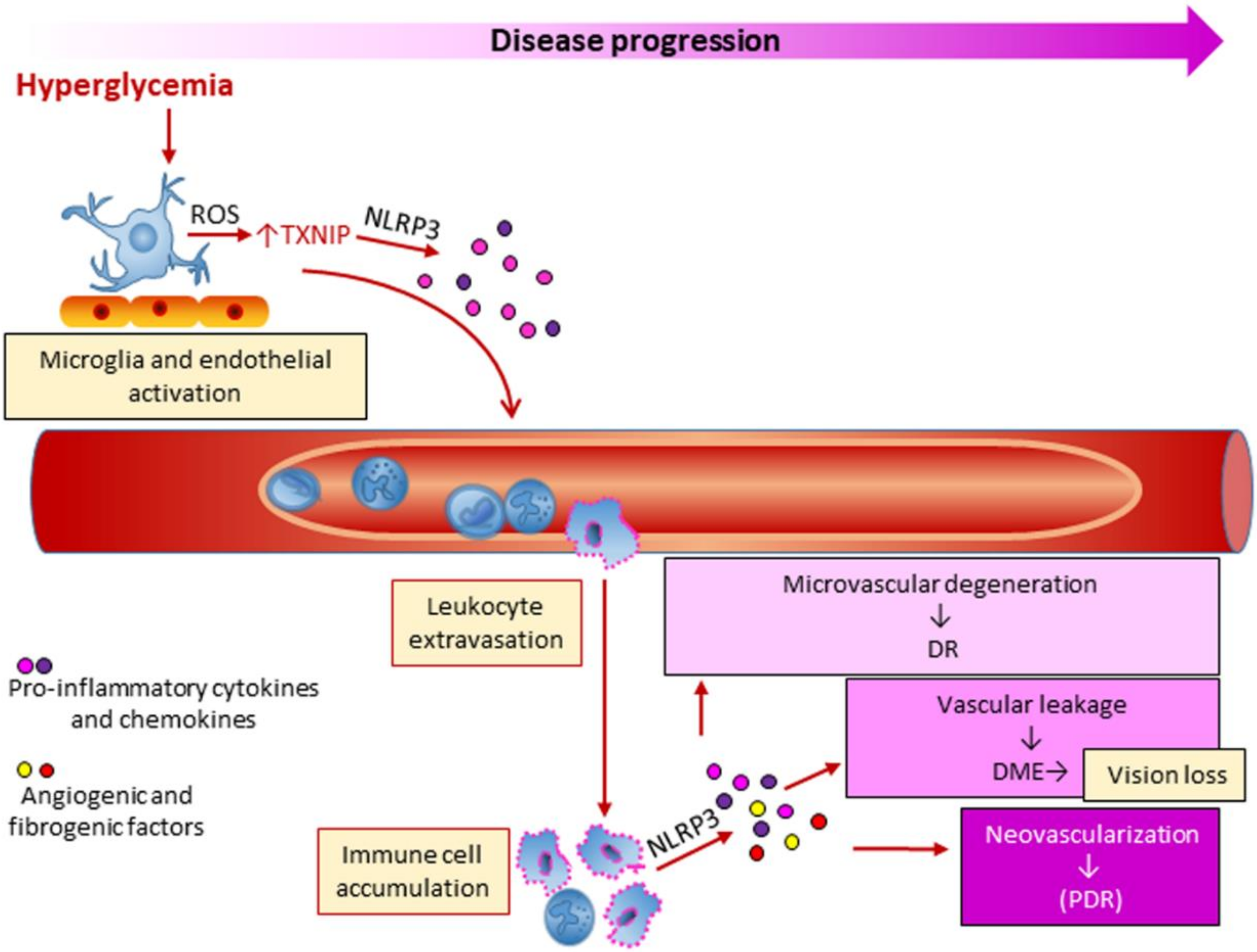
5. Diabetic Retinopathy
6. Diabetic Neuropathy
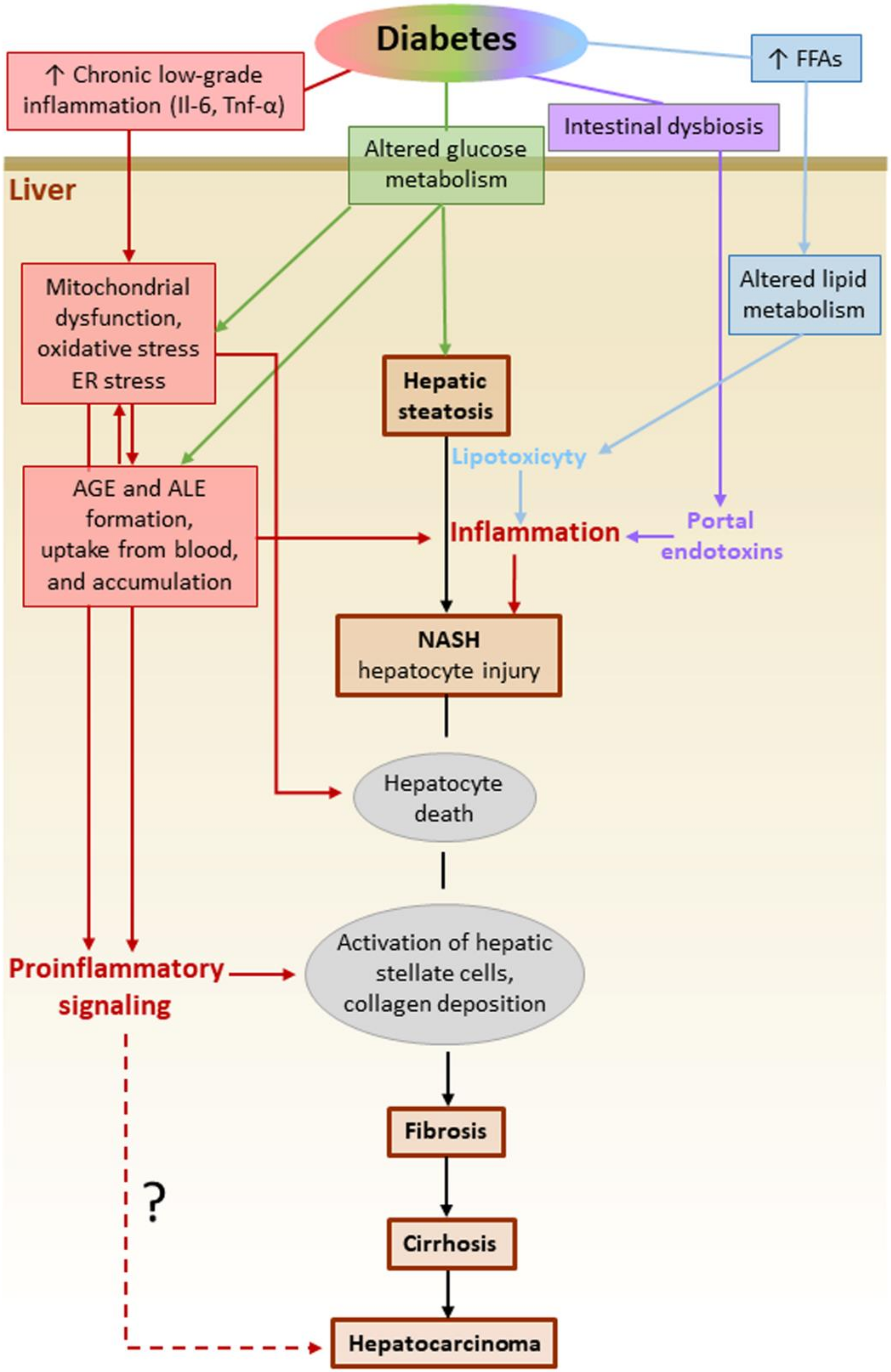
7. Diabetes-Associated Non-Alcoholic Fatty Liver Disease
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DM | diabetes mellitus |
| CVD | cardiovascular disease |
| IL | interleukin |
| NLR | NOD-like receptor |
| ADC | Apoptosis-associated speck-like protein containing a CARD |
| NLRPs | NLR proteins |
| HIN | interferon-inducible nature, and nuclear localization |
| AIM2 | absent in melanoma 2 |
| IFI16 | interferon γ-inducible protein 16 |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| TLRs | toll-like receptors |
| PRRs | pattern recognition receptors |
| PAMPS | pathogen- associated molecular patterns |
| DAMPs | damage-associated molecular patterns |
| NF-κB | nuclear factor-κB |
| ATP | adenosine triphosphate |
| ROS | reactive oxygen species |
| AGEs | advanced glycation end-products |
| FFAs | free fatty acids |
| MACEs | major adverse cardiovascular events |
| CANTOS | Canakinumab Anti-Inflammatory Thrombosis Outcomes Study |
| GLP | Glucagon-like peptide |
| SGLT | sodium glucose cotransporter |
| hsCRP | high-sensitivity C-reactive protein |
| CIRT | Cardiovascular Inflammation Reduction Trial |
| OxLDLs | oxidized LDLs |
| RAGE | receptor of AGEs |
| SREBP | sterol regulatory element binding protein |
| NETs | neutrophil extracellular traps |
| Foxp1 | forkhead box P transcription factor 1 |
| DKD | diabetic kidney disease |
| ESKD | end-stage kidney disease |
| GFR | glomerular filtration rate |
| RAS | renin-angiotensin system |
| KRIS | kidney risk inflammatory signature |
| TNF | tumor necrosis factor |
| BM | bone marrow |
| TXNIP | Thioredoxin Interacting Protein |
| PR2X7 | purinergic receptor 2X7 |
| DPP | dipeptidyl peptidase |
| DR | diabetic retinopathy |
| PDR | proliferative DR |
| STZ | streptozotocin |
| TRX1 | thioredoxin |
| ALEs | advanced lipoxidation endproducts |
| PGE2 | Prostaglandin E2 |
| EP2R | E-prostanoid receptor |
| PPARα | peroxisome proliferator-activated receptor-α |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
References
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Media Centre. Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 30 October 2018).
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, R.M.; Donath, M.Y.; Leroith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39, S244–S252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, K.R. Linking Metabolism and Immunology: Diabetic Nephropathy Is an Inflammatory Disease. J. Am. Soc. Nephrol. 2005, 16, 1537–1538. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Glaser, N.; Griffen, S.; Wang-Polagruto, J.; Miguelino, E.; Jialal, I. Increased Monocytic Activity and Biomarkers of Inflammation in Patients With Type 1 Diabetes. Diabetes 2006, 55, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes 2012, 62, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.; Komorowsky, C.V.; Weil, E.J.; Yee, B.; Hodgin, J.; Harder, J.L.; Godfrey, B.; Ju, W.; Boustany-Kari, C.M.; Schwarz, M.; et al. A molecular morphometric approach to diabetic kidney disease can link structure to function and outcome. Kidney Int. 2018, 93, 439–449. [Google Scholar] [CrossRef]
- Christgen, S.; Kanneganti, T.-D. Inflammasomes and the fine line between defense and disease. Curr. Opin. Immunol. 2020, 62, 39–44. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.C.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.L.; Stutz, A.; Alnemri, E.S.; Macdonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.E.; Fang, M.M.; Weinheimer-Haus, E.M.; Ennis, W.J.; Koh, T.J. Sustained Inflammasome Activity in Macrophages Impairs Wound Healing in Type 2 Diabetic Humans and Mice. Diabetes 2014, 63, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; De Marañon, A.M.; Iannantuoni, F.; López, E.-; López-Domènech, S.; Salom, C.; Jover, A.; Llabata, V.; Torres, I.R.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Poirier, H.; Shapiro, J.S.; Kim, R.J.; Lazar, M.A.; Amadio, L.; Oddi, G.; Ricci, C.; Pesce, C.; Pugliese, F.; et al. Deletion of p66Shc Longevity Gene Protects Against Experimental Diabetic Glomerulopathy by Preventing Diabetes-Induced Oxidative Stress. Diabetes 2006, 55, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Ricci, C.; Fantauzzi, C.B.; Pugliese, G. Protection from diabetes-induced atherosclerosis and renal disease by d-carnosine-octylester: Effects of early vs late inhibition of advanced glycation end-products in Apoe-null mice. Diabetologia 2014, 58, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N. Advanced glycation endproducts—role in pathology of diabetic complications. Diabetes Res. Clin. Pr. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus—Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, D.I.; Maahs, D.M.; Hovind, P.; Nakagawa, T. Uric Acid as a Mediator of Diabetic Nephropathy. Semin. Nephrol. 2011, 31, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, R.I.; Booth, J.W.; Mullins, J.J.; Bailey, M.A.; Tam, F.W.; Norman, J.T.; Unwin, R.J. Hyperglycemia-induced Renal P2X7 Receptor Activation Enhances Diabetes-related Injury. EBioMedicine 2017, 19, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solini, A.; Menini, S.; Rossi, C.; Ricci, C.; Santini, E.; Fantauzzi, C.B.; Iacobini, C.; Pugliese, G. The purinergic 2X7receptor participates in renal inflammation and injury induced by high-fat diet: Possible role of NLRP3 inflammasome activation. J. Pathol. 2013, 231, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Fantauzzi, C.B.; Pesce, C.; Giaccari, A.; Salomone, E.; Lapolla, A.; Orioli, M.; Aldini, G.; Pugliese, G. FL-926-16, a novel bioavailable carnosinase-resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy indb/dbmice. Br. J. Pharmacol. 2017, 175, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.; Hwang, I.; Han, S.H.; Shin, J.-S.; Shin, O.S.; Yu, J.-W. Advanced glycation end products impair NLRP3 inflammasome–mediated innate immune responses in macrophages. J. Biol. Chem. 2017, 292, 20437–20448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejnovic, N.; Pantic, J.; Jovanovic, I.; Radosavljevic, G.; Milovanovic, M.; Nikolić, I.; Zdravkovic, N.; Djukic, A.; Arsenijević, N.N.; Lukic, M.L. Galectin-3 Deficiency Accelerates High-Fat Diet–Induced Obesity and Amplifies Inflammation in Adipose Tissue and Pancreatic Islets. Diabetes 2013, 62, 1932–1944. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef]
- American Diabetes Association Addendum. 10. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; E Grobbee, D.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Hear. J. 2019, 41, 255–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Prato, S. Megatrials in type 2 diabetes. From excitement to frustration? Diabetol. 2009, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Furtado, R.H.M.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; et al. Comparison of the effects of glucagon-like peptide receptor agonists and sodium-glucose cotransporter 2 inhibitors for prevention of major adverse cardiovascular and renal outcomes in type 2 diabetes mellitus: Systematic review and meta-analysis of cardiovascular outcomes trials. Circulation 2019, 139, 2022–2031. [Google Scholar]
- Kanter, J.E.; Averill, M.M.; Leboeuf, R.C.; Bornfeldt, K.E. Diabetes-accelerated atherosclerosis and inflammation. Circ. Res. 2008, 103, e116–e117. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Kastelein, J.; Koenig, W.; Genest, J.; Lorenzatti, A.; et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. New Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wad, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Rader, D. IL-1 and atherosclerosis: A murine twist to an evolving human story. J. Clin. Investig. 2011, 122, 27–30. [Google Scholar] [CrossRef]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.-G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. New Engl. J. Med. 2018, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 Gene Silencing Ameliorates Diabetic Cardiomyopathy in a Type 2 Diabetes Rat Model. PLOS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, G.; Alarcón, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.V.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepúlveda, M.; et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klen, J.; Goričar, K.; Janež, A.; Dolžan, V. NLRP3 Inflammasome Polymorphism and Macrovascular Complications in Type 2 Diabetes Patients. J. Diabetes Res. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.X.; Ling, S.K.; Hu, M.Y.; Ma, Y.; Li, Y.; Huang, P.L. Protective Effects of Acarbose Against Vascular Endothelial Dysfunction through Inhibiting Nox4/NLRP3 Inflammasome Pathway in Diabetic Rats. Free Radic. Biol. Med. 2019, 145, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ko, Y.S.; Park, S.W.; Kim, H.J.; Par, S.W. P2Y2R activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free. Radic. Biol. Med. 2019, 136, 109–117. [Google Scholar] [CrossRef]
- Wang, R.; Wu, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Li, W.; Wang, S. Activation of NLRP3 Inflammasome Promotes Foam Cell Formation in Vascular Smooth Muscle Cells and Atherogenesis Via HMGB1. J. Am. Hear. Assoc. 2018, 7, 008596. [Google Scholar] [CrossRef] [Green Version]
- Chellan, B.; Reardon, C.A.; Getz, G.S.; Bowman, M.A.H. Enzymatically Modified Low-Density Lipoprotein Promotes Foam Cell Formation in Smooth Muscle Cells via Macropinocytosis and Enhances Receptor-Mediated Uptake of Oxidized Low-Density Lipoprotein. Arter. Thromb. Vasc. Biol. 2016, 36, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Saremi, A.; Howell, S.; Schwenke, D.C.; Bahn, G.; Beisswenger, P.J.; Reaven, P.D. Advanced Glycation End Products, Oxidation Products, and the Extent of Atherosclerosis During the VA Diabetes Trial and Follow-up Study. Diabetes Care 2017, 40, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, S.; Jiang, B.; Cohen, R.A.; Zang, M. Activation of Sterol Regulatory Element Binding Protein and NLRP3 Inflammasome in Atherosclerotic Lesion Development in Diabetic Pigs. PLOS ONE 2013, 8, e67532. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Hear. Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Avondt, K.; Maegdefessel, L.; Soehnlein, O. Therapeutic Targeting of Neutrophil Extracellular Traps in Atherogenic Inflammation. Thromb. Haemost. 2019, 119, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Pereira, C.A.; Carlos, D.; Ferreira, N.S.; Silva, J.F.; Zanotto, C.Z.; Zamboni, D.S.; Garcia, V.D.; Ventura, D.F.; Silva, J.S.; Tostes, R.C. Mitochondrial DNA Promotes NLRP3 Inflammasome Activation and Contributes to Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Front. Physiol. 2020, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- A Zelniker, T.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; De Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988–2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef]
- Saran, R.; Li, Y.; Robinson, B.; Ayanian, J.; Balkrishnan, R.; Bragg-Gresham, J.; Chen, J.T.; Cope, E.; Gipson, D.; He, K.; et al. US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2015, 66, S1–S305. [Google Scholar] [CrossRef] [Green Version]
- Reutens, A.T. Epidemiology of Diabetic Kidney Disease. Med. Clin. North Am. 2013, 97, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, A.A.; Segel, J.E.; Zhuo, X.; Hoerger, T.J.; Imai, K.; Williams, D. Medical Costs of CKD in the Medicare Population. J. Am. Soc. Nephrol. 2013, 24, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Christensen, C.K.; Vittinghus, E. The Stages in Diabetic Renal Disease: With Emphasis on the Stage of Incipient Diabetic Nephropathy. Diabetes 1983, 32, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G. Updating the natural history of diabetic nephropathy. Acta Diabetol. 2014, 51, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Italian Diabetes Society and the Italian Society of Nephrology; Penno, G.; Natali, A.; Barutta, F.; Di Paolo, S.; Reboldi, G.; Gesualdo, L.; De Nicola, L. Diabetic kidney disease: New clinical and therapeutic issues. Joint position statement of the Italian Diabetes Society and the Italian Society of Nephrology on “The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function”. J. Nephrol. 2019, 33, 9–35. [Google Scholar] [CrossRef] [Green Version]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T. Revisiting inflammation in diabetic nephropathy: The role of the Nlrp3 inflammasome in glomerular resident cells. Kidney Int. 2015, 87, 12–14. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Dom, Z.I.M.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.-J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF Receptors 1 and 2 Predict ESRD in Type 2 Diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Furuta, T.; Saito, T.; Ootaka, T.; Soma, J.; Obara, K.; Abe, K.; Yoshinaga, K. The Role of Macrophages in Diabetic Glomerulosclerosis. Am. J. Kidney Dis. 1993, 21, 480–485. [Google Scholar] [CrossRef]
- Viedt, C.; Orth, S. Monocyte chemoattractant protein-1 (MCP-1) in the kidney: Does it more than simply attract monocytes? Nephrol. Dial. Transplant. 2002, 17, 2043–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Zheng, B.; Cleary, C.M.; Ruiz-Ortega, M.; Ortiz, A.; Fernandez-Fernandez, B. Targeting inflammation in diabetic kidney disease: Early clinical trials. Expert Opin. Investig. Drugs 2016, 25, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Alexandraki, K.I.; Piperi, C.; Kalofoutis, C.; Singh, J.; Alaveras, A.; Kalofoutis, A. Inflammatory Process in Type 2 Diabetes: The Role of Cytokines. Ann. New York Acad. Sci. 2006, 1084, 89–117. [Google Scholar] [CrossRef]
- Nakamura, A.; Shikata, K.; Hiramatsu, M.; Nakatou, T.; Kitamura, T.; Wada, J.; Itoshima, T.; Makino, H. Serum Interleukin-18 Levels Are Associated With Nephropathy and Atherosclerosis in Japanese Patients With Type 2 Diabetes. Diabetes Care 2005, 28, 2890–2895. [Google Scholar] [CrossRef] [Green Version]
- Stokman, G.; Kers, J.; Ünsal, Y.; Hoelbeek, J.; Claessen, N.; De Boer, O.J.; Netea, M.G.; Hilbrands, L.; Bemelman, F.J.; Berge, I.J.T.; et al. Predominant Tubular Interleukin-18 Expression in Polyomavirus-Associated Nephropathy. Transplant. 2016, 100, e88–e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Yuan, J.; Zhao, Y.; Zha, Y. Urine interleukin-18 in prediction of acute kidney injury: A systemic review and meta-analysis. J. Nephrol. 2014, 28, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Kohli, S.; Nazir, S.; Ghosh, S.; Ranjan, S.; Wang, H.; Madhusudhan, T.; et al. Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Li, G.; Chen, Y.; Conley, S.M.; Gehr, T.W.B.; Boini, K.M.; Li, P.-L. Nod-like Receptor Protein 3 (NLRP3) Inflammasome Activation and Podocyte Injury via Thioredoxin-Interacting Protein (TXNIP) during Hyperhomocysteinemia. J. Biol. Chem. 2014, 289, 27159–27168. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rodriguez, S.; Caballo, C.; Gutierrez, G.; Vera, M.; Cruzado, J.M.; Cases, A.; Escolar, G.; Diaz-Ricart, M. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur. J. Clin. Investig. 2015, 45, 160–169. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-Independent NLRP3 Augments TGF-β Signaling in Kidney Epithelium. J. Immunol. 2012, 190, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wen, Y.; Tang, T.-T.; Lv, L.-L.; Tang, R.-N.; Liu, H.; Ma, K.-L.; Crowley, S.D.; Liu, B.-C. Megalin/Cubulin-Lysosome-mediated Albumin Reabsorption Is Involved in the Tubular Cell Activation of NLRP3 Inflammasome and Tubulointerstitial Inflammation. J. Biol. Chem. 2015, 290, 18018–18028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; Correia, J.D.S.; Ulevitch, R.J.; Hoffman, H.M.; McKay, D.B. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Xu, M.; Ding, L.-H.; Lv, L.-L.; Liu, H.; Ma, K.-L.; Zhang, A.-H.; Crowley, S.D.; Liu, B. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int. J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Lee, S.-H.; Kim, Y.-G.; Kim, S.-Y.; Seo, J.-W.; Choi, Y.-W.; Kim, D.-J.; Jeong, K.-H.; Lee, T.; Ihm, C.-G.; et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am. J. Physiol. Physiol. 2015, 308, F993–F1003. [Google Scholar] [CrossRef]
- Darisipudi, M.N.; Thomasova, D.; Mulay, S.R.; Brech, R.; Noessner, E.; Liapis, H.; Anders, H.-J. Uromodulin Triggers IL-1β–Dependent Innate Immunity via the NLRP3 Inflammasome. J. Am. Soc. Nephrol. 2012, 23, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Allam, R.; Darisipudi, M.N.; Tschopp, J.; Anders, H.-J. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur. J. Immunol. 2013, 43, 3336–3342. [Google Scholar] [CrossRef]
- Prencipe, G.; Caiello, I.; Cherqui, S.; Whisenant, T.; Petrini, S.; Emma, F.; De Benedetti, F. Inflammasome Activation by Cystine Crystals: Implications for the Pathogenesis of Cystinosis. J. Am. Soc. Nephrol. 2014, 25, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S.; et al. High Glucose and Lipopolysaccharide Prime NLRP3 Inflammasome via ROS/TXNIP Pathway in Mesangial Cells. J. Diabetes Res. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Liu, S.; Wang, H.; Dou, H. Role of the thioredoxin interacting protein in diabetic nephropathy and the mechanism of regulating NOD-like receptor protein 3 inflammatory corpuscle. Int. J. Mol. Med. 2019, 43, 2440–2450. [Google Scholar] [CrossRef]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S.; et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Peng, R.; Zhang, L.-Y.; Sun, Y.; Peng, H.-M.; Liu, H.-D.; Yu, L.-J.; Li, A.-L.; Zhang, Y.-J.; Jiang, W.-H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2583. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Natarajan, R. Long Noncoding RNAs in Diabetes and Diabetic Complications. Antioxidants Redox Signal. 2018, 29, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Leti, F.; Distefano, J.K. Long Noncoding RNAs as Diagnostic and Therapeutic Targets in Type 2 Diabetes and Related Complications. Genes 2017, 8, 207. [Google Scholar] [CrossRef] [Green Version]
- Raut, S.K.; Khullar, M. The Big Entity of New RNA World: Long Non-Coding RNAs in Microvascular Complications of Diabetes. Front. Endocrinol. 2018, 9, 300. [Google Scholar] [CrossRef]
- Fantauzzi, C.B.; Menini, S.; Iacobini, C.; Rossi, C.; Santini, E.; Solini, A.; Pugliese, G. Deficiency of the Purinergic Receptor 2X7 Attenuates Nonalcoholic Steatohepatitis Induced by High-Fat Diet: Possible Role of the NLRP3 Inflammasome. Oxidative Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Li, G.; Zhang, Q.; Ritter, J.; Li, W.; Li, P.-L. D-Ribose Induces Podocyte NLRP3 Inflammasome Activation and Glomerular Injury via AGEs/RAGE Pathway. Front. Cell Dev. Biol. 2019, 7, 259. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Bajaj, M.; Qian, J.; Ye, Y. Dipeptidyl peptidase-4 inhibition by Saxagliptin prevents inflammation and renal injury by targeting the Nlrp3/ASC inflammasome. BMJ Open Diabetes Res. Care 2016, 4, e000227. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Bajaj, M.; Yang, H.C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLOS ONE 2017, 12, e0181248. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Nazir, S.; Wang, H.; Kohli, S.; Ranjan, S.; Mertens, P.R.; Nawroth, P.P.; et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 2016, 6, 34228. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yin, N.; Liu, W.; Cui, X.; Chen, S.; Wang, E. Curcumin Ameliorates Diabetic Nephropathy by Suppressing NLRP3 Inflammasome Signaling. BioMed Res. Int. 2017, 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P.; et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Liu, X.; Yin, J.; Wu, H.; Cai, X.; Wang, N.; Qian, Y.; Wang, F. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism 2018, 83, 18–24. [Google Scholar] [CrossRef]
- Yang, S.-M.; Ka, S.-M.; Wu, H.-L.; Yeh, Y.-C.; Kuo, C.-H.; Hua, K.-F.; Shi, G.-Y.; Hung, Y.-J.; Hsiao, F.-C.; Yang, S.-S.; et al. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia 2013, 57, 424–434. [Google Scholar] [CrossRef]
- Soares, J.L.; Fernandes, F.P.; Patente, T.A.; Monteiro, M.B.; Parisi, M.C.; Giannella-Neto, D.; Correa-Giannella, M.L.; Pontillo, A. Gain-of-function variants in NLRP1 protect against the development of diabetic kidney disease: NLRP1 inflammasome role in metabolic stress sensing? Clin. Immunol. 2018, 187, 46–49. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.G.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Prim. 2016, 2, 16013. [Google Scholar] [CrossRef]
- Richter, C.U. Management of diabetic retinopathy. In Primary Care Medicine; Gorrol, A.H., May, L.A., Mulley, A.G., Jr., Eds.; J.P. Lippincott Company: London, UK, 1987; pp. 857–858. [Google Scholar]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, 93751. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.J.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.-P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Schröder, S.; Palinski, W.; Schmid-Schönbein, G.W. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am. J. Pathol. 1991, 139, 81–100. [Google Scholar]
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Loukovaara, S.; Nurkkala, H.; Tamene, F.; Gucciardo, E.; Liu, X.; Repo, P.; Lehti, K.; Varjosalo, M. Quantitative Proteomics Analysis of Vitreous Humor from Diabetic Retinopathy Patients. J. Proteome Res. 2015, 14, 5131–5143. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-Y.; Green, W.R.; Tso, M.O.M. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhao, M.; Zhao, S.; Lu, Q.; Ni, L.; Zou, C.; Lu, L.; Xu, X.; Guan, H.; Zheng, Z.; et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: A novel inhibitory effect of minocycline. Inflamm. Res. 2016, 66, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lv, X.; Hu, Z.; Ye, X.; Zheng, X.; Ding, Y.; Xie, P.; Liu, Q. Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis. 2017, 8, e2941. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Lee, I.; Hüttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP Links Innate Host Defense Mechanisms to Oxidative Stress and Inflammation in Retinal Muller Glia under Chronic Hyperglycemia: Implications for Diabetic Retinopathy. Exp. Diabetes Res. 2012, 2012, 1–19. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [Green Version]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients With Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2017, 58, 5594–5603. [Google Scholar] [CrossRef]
- Wu, H.; Hwang, D.-K.; Song, X.; Tao, Y. Association between Aqueous Cytokines and Diabetic Retinopathy Stage. J. Ophthalmol. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Loukovaara, S.; Piippo, N.; Kinnunen, K.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017, 95, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, R.; Bligard, G.W.; Zhang, S.; Yin, L.; Lukasiewicz, P.; Semenkovich, C.F. Functional Deficits Precede Structural Lesions in Mice With High-Fat Diet–Induced Diabetic Retinopathy. Diabetes 2016, 65, 1072–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnick, G.H.; Korth, K.; Groo, A.; Palta, M. Electroretinographic Oscillatory Potentials Predict Progression of Diabetic Retinopathy. Arch. Ophthalmol. 1984, 102, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alcalde-Estevez, E.; García-Ramírez, M.; Cazzoni, D.; De La Villa, P.; Sánchez-Fernández, E.M.; Mellet, C.O.; García-Fernández, J.M.; Hernández, C.; Simó, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db / db mice. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, X.; Liao, N.; Mi, L.; Peng, Y.; Liu, B.; Zhang, S.; Wen, F. Enhanced Expression of NLRP3 Inflammasome-Related Inflammation in Diabetic Retinopathy. Investig. Opthalmology Vis. Sci. 2018, 59, 978–985. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, S.B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A.; et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci. Rep. 2018, 8, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, C.; Wan, X.-S.; Li, S.-W. NLRP1 deficiency attenuates diabetic retinopathy (DR) in mice through suppressing inflammation response. Biochem. Biophys. Res. Commun. 2018, 501, 351–357. [Google Scholar] [CrossRef]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells—Implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2009, 11, 136–140. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Montemari, A.; Powell, F.L.; Malla, P.; Gutsaeva, D.R.; Bachettoni, A.; Ripandelli, G.; Repossi, A.; Tawfik, A.; Martin, P.M.; et al. Monosodium Urate Contributes to Retinal Inflammation and Progression of Diabetic Retinopathy. Diabetes 2019, 68, 1014–1025. [Google Scholar] [CrossRef] [Green Version]
- Platania, C.B.M.; Giurdanella, G.; Di Paola, L.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem. Pharmacol. 2017, 138, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lu, Q.; Chen, W.; Li, J.; Li, C.; Zheng, Z. Vitamin D3 Protects against Diabetic Retinopathy by Inhibiting High-Glucose-Induced Activation of the ROS/TXNIP/NLRP3 Inflammasome Pathway. J. Diabetes Res. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wang, Y.; Xie, T.; Zhan, P.; Zou, J.; Nie, X.; Shao, J.; Zhuang, M.; Tan, C.; Tan, J.; et al. Prostaglandin E2/EP2 receptor signalling pathway promotes diabetic retinopathy in a rat model of diabetes. Diabetologia 2019, 62, 335–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, A.C.; Mott, R.; Lyons, T.J.; Ma, J.-X. Therapeutic Effects of PPARα Agonists on Diabetic Retinopathy in Type 1 Diabetes Models. Diabetes 2012, 62, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, Y.; Ding, L.; He, X.; Takahashi, Y.; Gao, Y.; Shen, W.; Cheng, R.; Chen, Q.; Qi, X.; et al. Pathogenic role of diabetes-induced PPAR- down-regulation in microvascular dysfunction. Proc. Natl. Acad. Sci. 2013, 110, 15401–15406. [Google Scholar] [CrossRef] [Green Version]
- Noonan, J.E.; Jenkins, A.J.; Ma, J.-X.; Keech, A.C.; Wang, J.J.; Lamoureux, E.L. An Update on the Molecular Actions of Fenofibrate and Its Clinical Effects on Diabetic Retinopathy and Other Microvascular End Points in Patients With Diabetes. Diabetes 2013, 62, 3968–3975. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.; Mitchell, P.; Summanen, P.; O’Day, J.; Davis, T.; Moffitt, M.; Taskinen, M.-R.; Simes, R.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- The ACCORD Study Group and ACCORD Eye Study Group; ACCORD Study Group; ACCORD Eye Study Group; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; et al. Effects of Medical Therapies on Retinopathy Progression in Type 2 Diabetes. New Engl. J. Med. 2010, 363, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, F.; Zhang, X.; Cheng, R.; Ma, J.-X.; Yi, J.; Li, J. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol. Cell. Biochem. 2017, 445, 105–115. [Google Scholar] [CrossRef]
- Vincent, A.M.; Callaghan, B.C.; Smith, A.L.; Feldman, E. Diabetic neuropathy: Cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 2011, 7, 573–583. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, C.; Yan, B.; Shi, X.; Shi, Y.; Qu, L.; Liang, X. Jinmaitong Ameliorates Diabetic Peripheral Neuropathy Through Suppressing TXNIP/NLRP3 Inflammasome Activation In The Streptozotocin-Induced Diabetic Rat Model. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 2145–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Lin, X.; Guan, M.; Zeng, Y.; Liu, Y. Verapamil Attenuated Prediabetic Neuropathy in High-Fat Diet-Fed Mice through Inhibiting TXNIP-Mediated Apoptosis and Inflammation. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Huang, Y.; Zhou, F.; Zhao, B.; Zhongyuan, X. Dexmedetomidine attenuates P2X4 and NLRP3 expression in the spine of rats with diabetic neuropathic pain. Acta Cir. Bras. 2019, 34. [Google Scholar] [CrossRef] [Green Version]
- Thakur, V.; Sadanandan, J.; Chattopadhyay, M. High-Mobility Group Box 1 Protein Signaling in Painful Diabetic Neuropathy. Int. J. Mol. Sci. 2020, 21, 881. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Dai, J.; Li, L.; Chen, H.; Chai, Y. NLRP3 Inflammasome Expression and Signaling in Human Diabetic Wounds and in High Glucose Induced Macrophages. J. Diabetes Res. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Noureddin, M.; Rinella, M.E. Nonalcoholic Fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin. Liver Dis. 2015, 19, 361–379. [Google Scholar] [CrossRef]
- Puri, P.; Sanyal, A.J. Nonalcoholic fatty liver disease: Definitions, risk factors, and workup. Clin. Liver Dis. 2012, 1, 99–103. [Google Scholar] [CrossRef]
- Howard, W. Diagnosis and Management of the Metabolic Syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Yearb. Endocrinol. 2006, 2006, 113–114. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Berk, P.D. Lipid metabolism in hepatic steatosis. Clin. Liver Dis. 2004, 8, 639–671. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef] [Green Version]
- Muraki, Y.; Makita, Y.; Yamasaki, M.; Amano, Y.; Matsuo, T. Elevation of liver endoplasmic reticulum stress in a modified choline-deficient l -amino acid-defined diet-fed non-alcoholic steatohepatitis mouse model. Biochem. Biophys. Res. Commun. 2017, 486, 632–638. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Stitt, A.W.; Gardiner, T.A.; Anderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE Inhibitor Pyridoxamine Inhibits Development of Retinopathy in Experimental Diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free. Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Menini, S.; Ricci, C.; Fantauzzi, C.B.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef]
- Di Maira, G.; Pastore, M.; Marra, F. Liver fibrosis in the context of nonalcoholic steatohepatitis: The role of adipokines. Minerva Gastroenterol. e Dietol. 2017, 64, 39–50. [Google Scholar]
- Carnevale, R.; Pastori, D.; Nocella, C.; Cammisotto, V.; Baratta, F.; Del Ben, M.; Angelico, F.; Sciarretta, S.; Bartimoccia, S.; Novo, M.; et al. Low-grade endotoxemia, gut permeability and platelet activation in patients with impaired fasting glucose. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. Liver Physiol. 2011, 302, G77–G84. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; A Brenner, D.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Pillai, A.; Ganz, M.; Lippai, R.; Petrášek, J.; Park, J.-K.; Kodys, K.; Dolganiuc, A.; Kurt-Jones, E.A.; Szabo, S.B.G. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014, 34, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- Hertog, M.G.; Hollman, P.C. Potential health effects of the dietary flavonol quercetin. Eur. J. Clin. Nutr. 1996, 50, 63–71. [Google Scholar] [PubMed]
- Spahr, L.; Amann, P.; Kern, I.; Golaz, O.; Frossard, J.-L.; Hadengue, A.; Bresson-Hadni, S. Allopurinol, oxidative stress and intestinal permeability in patients with cirrhosis: An open-label pilot study. Liver Int. 2007, 27, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, C.; Ding, X.-Q.; Pan, Y.; Gu, T.-T.; Wang, M.-X.; Liu, Y.-L.; Wang, F.-M.; Wang, S.-J.; Kong, L.-D. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to alleviate inflammation and lipid accumulation in diabetic rats. Br. J. Pharmacol. 2013, 169, 1352–1371. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetol. 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Gao, X.; Fan, J.-G. Diagnosis and management of non-alcoholic fatty liver disease and related metabolic disorders: Consensus statement from the Study Group of Liver and Metabolism, Chinese Society of Endocrinology. J. Diabetes 2013, 5, 406–415. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; A Aldersley, M.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Jinnouchi, H.; Sugiyama, S.; Yoshida, A.; Hieshima, K.; Kurinami, N.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; Matsui, K.; Jinnouchi, T. Liraglutide, a Glucagon-Like Peptide-1 Analog, Increased Insulin Sensitivity Assessed by Hyperinsulinemic-Euglycemic Clamp Examination in Patients with Uncontrolled Type 2 Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q.-L. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Mridha, A.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, F.; Wang, Y.; Gisterå, A.; Roy, J.; Paulsson-Berne, G.; Hedin, U.; Lerman, A.; Hansson, G.K.; Herrmann, J.; et al. Inflammasome-Driven Interleukin-1α and Interleukin-1β Production in Atherosclerotic Plaques Relates to Hyperlipidemia and Plaque Complexity. JACC Basic Transl. Sci. 2019, 4, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menini, S.; Iacobini, C.; Vitale, M.; Pugliese, G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells 2020, 9, 1812. https://doi.org/10.3390/cells9081812
Menini S, Iacobini C, Vitale M, Pugliese G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells. 2020; 9(8):1812. https://doi.org/10.3390/cells9081812
Chicago/Turabian StyleMenini, Stefano, Carla Iacobini, Martina Vitale, and Giuseppe Pugliese. 2020. "The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders" Cells 9, no. 8: 1812. https://doi.org/10.3390/cells9081812
APA StyleMenini, S., Iacobini, C., Vitale, M., & Pugliese, G. (2020). The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells, 9(8), 1812. https://doi.org/10.3390/cells9081812