Caenorhabditis elegans Deficient in DOT-1.1 Exhibit Increases in H3K9me2 at Enhancer and Certain RNAi-Regulated Regions


Abstract
:1. Introduction
2. Materials and Methods
2.1. C. elegans Growth Conditions and Strains
2.2. Evaluation of Gonad Migration Defects
2.3. Chromatin Immunoprecipitation
2.4. H3K9me2 ChIP-seq Data Pre-Processing and Read Mapping
2.5. Coordinates of Enhancers, ATAC-seq Peaks, Promoters, ZFP-1 Peaks, ALG-3/4 Targets, Genomic Repeats, Chromosome Domains, and ERGO-1/RRF-3 Targets
2.6. Comparison of Cumulative H3K9me2 and ZFP-1 ChIP-seq Signals at Genomic Regions and at Enhancer Regions
2.7. H3K9me2 ChIP-seq Data Analysis
3. Results
3.1. H3K9me2 and ZFP-1/DOT-1.1 Occupy Different Genomic Locations
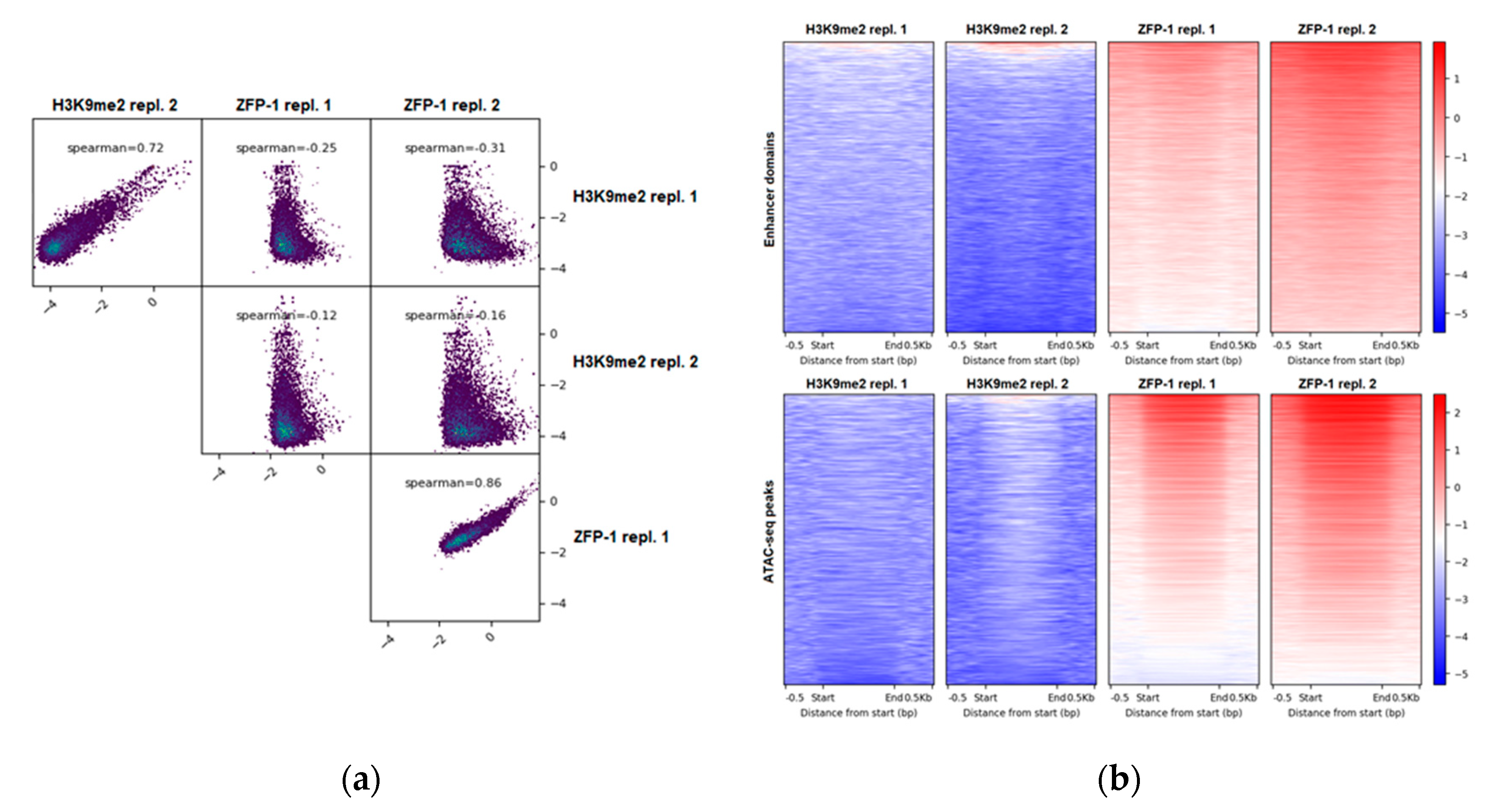
3.1.1. Genome-Wide Anti-Correlation
3.1.2. H3K9me2 and ZFP-1 at Developmental Enhancers
3.2. DOT-1.1 Loss Does Not Perturb the Preferential Localization of H3K9me2 to Autosome Arms
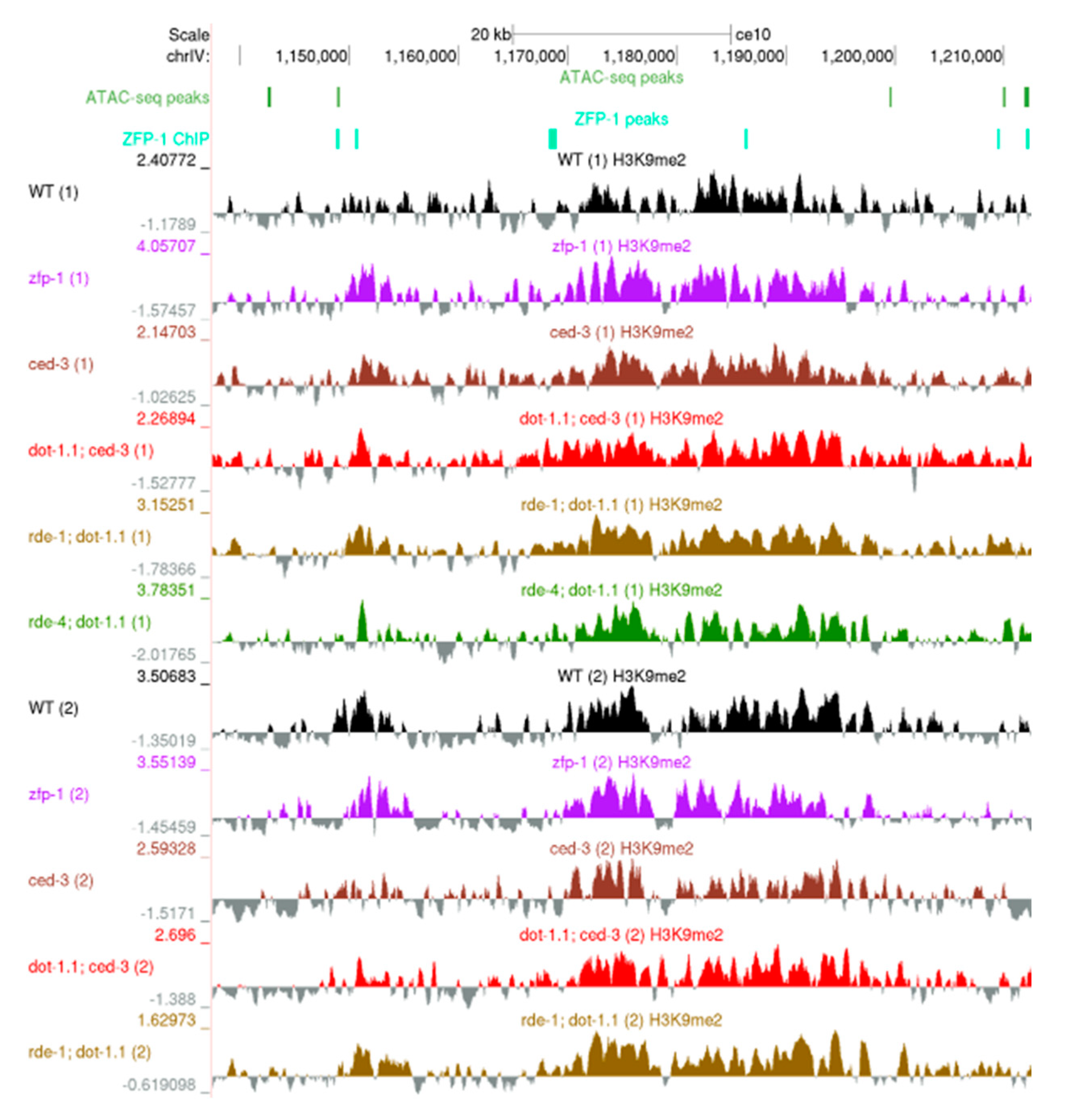
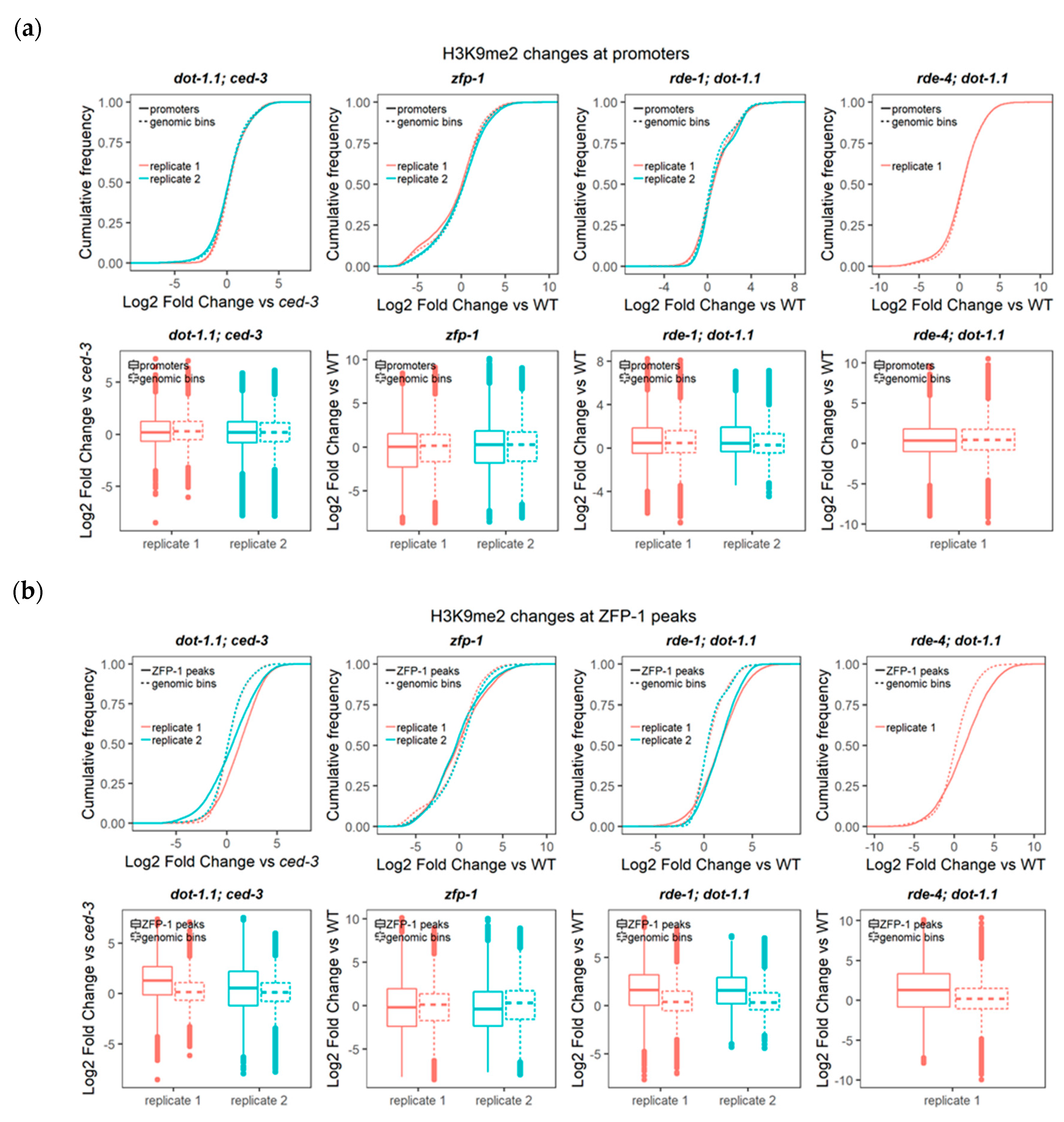
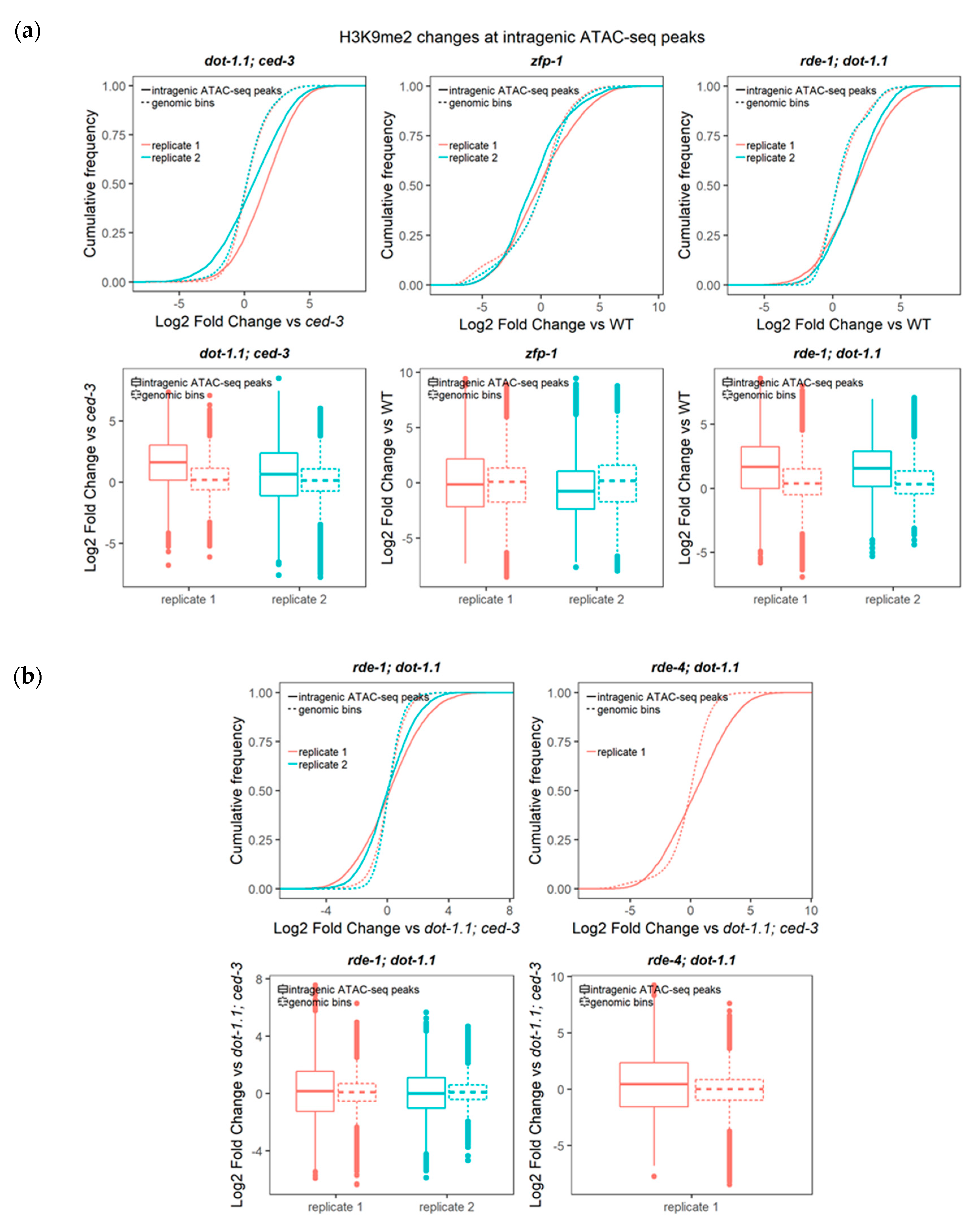
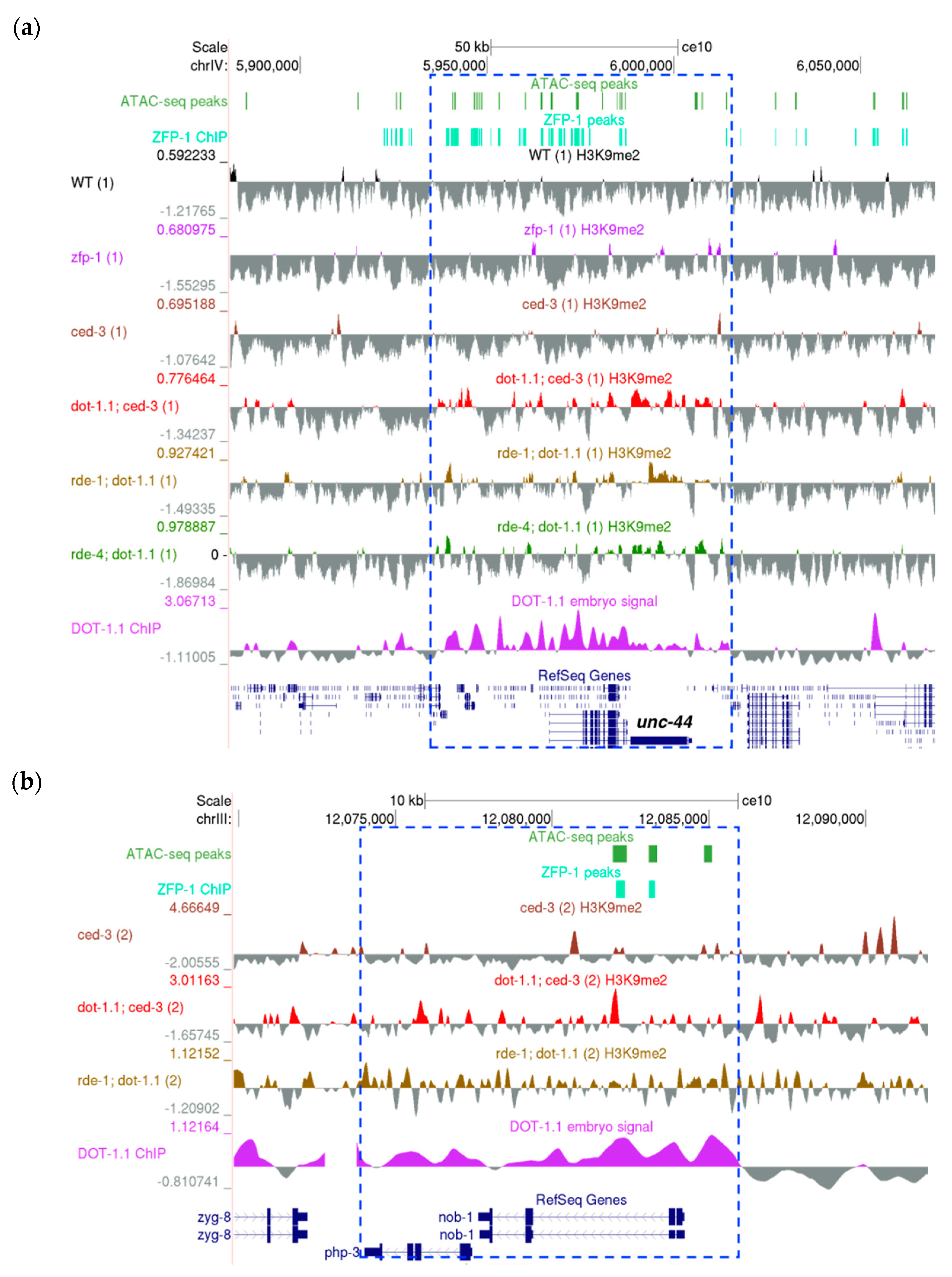
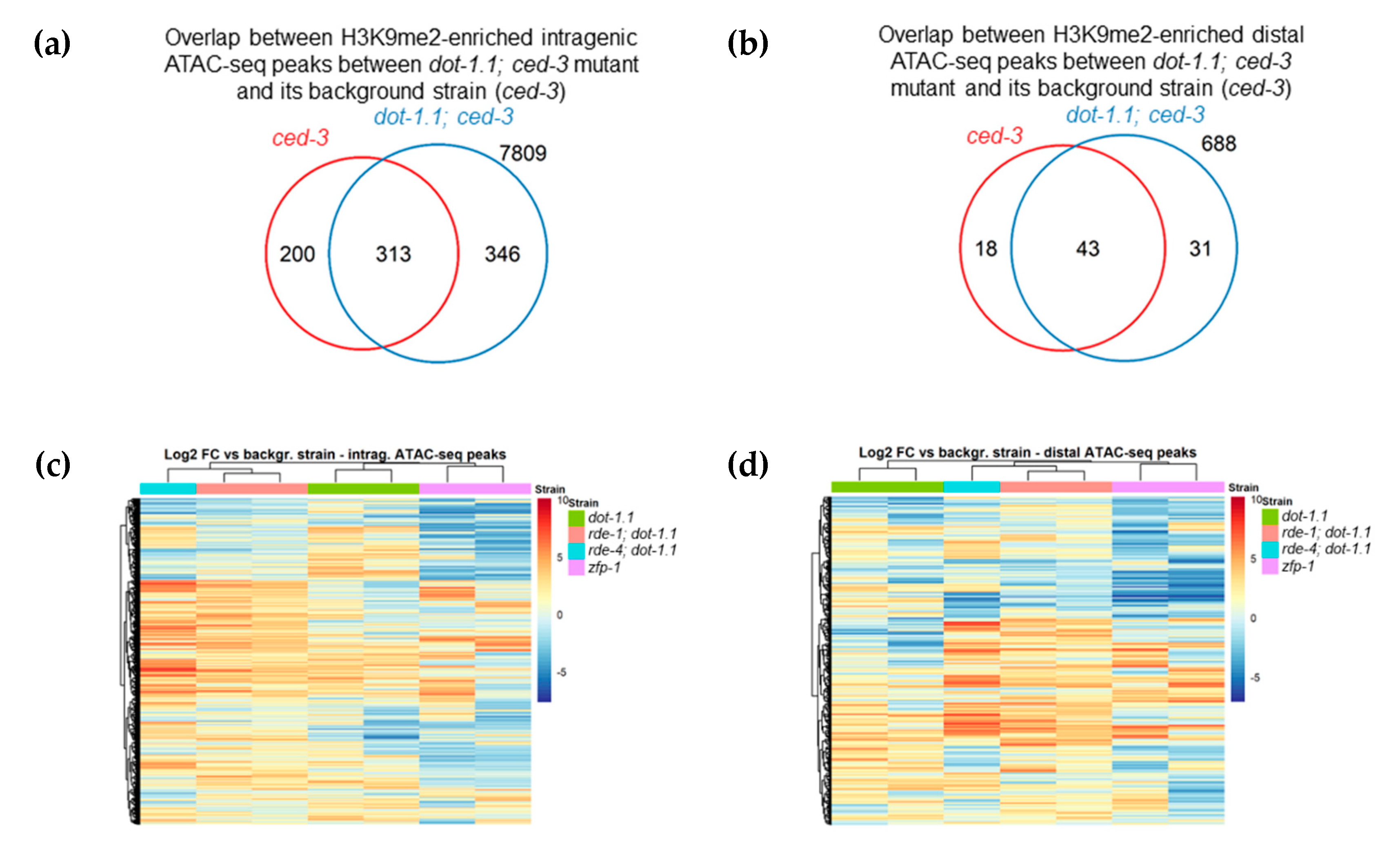
3.3. Regions Corresponding to ZFP-1 Peaks, But Not Promoters, Gain H3K9me2 upon DOT-1.1 Loss
3.4. H3K9me2 Is Elevated at Enhancers in dot-1.1(−) Worms
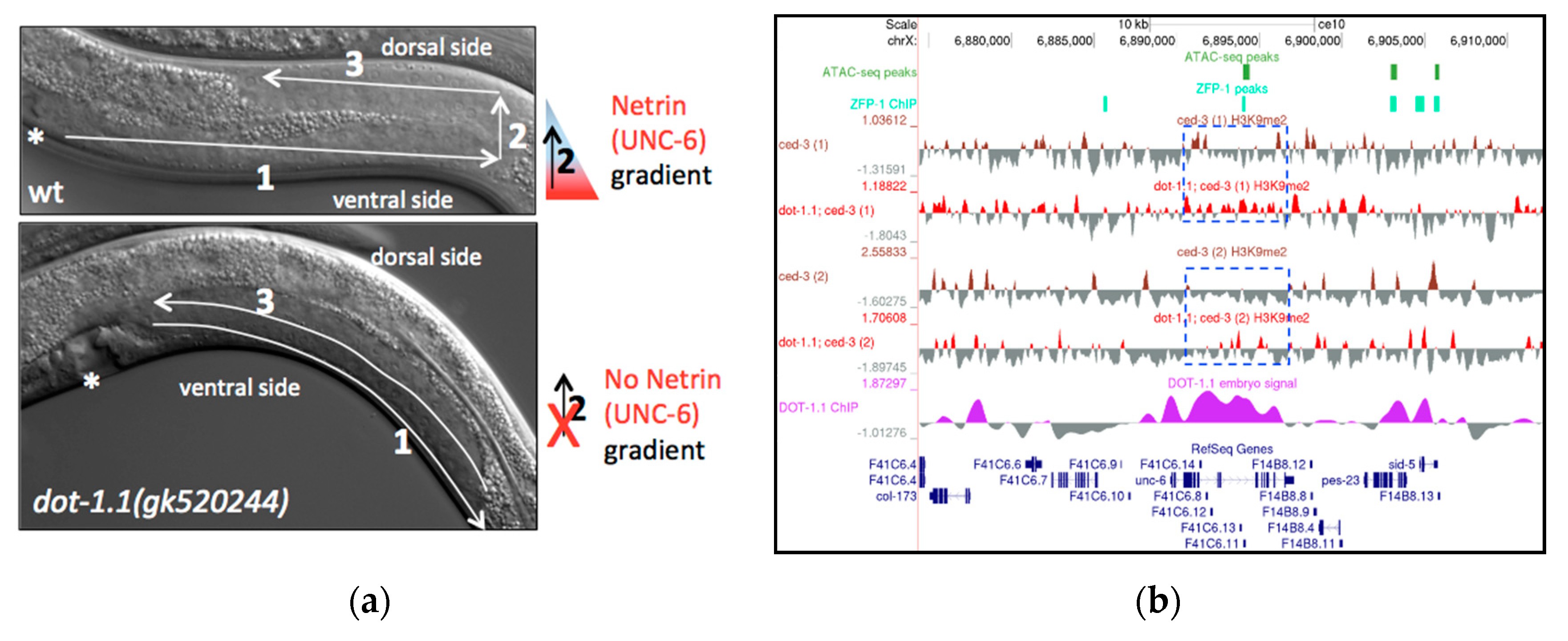
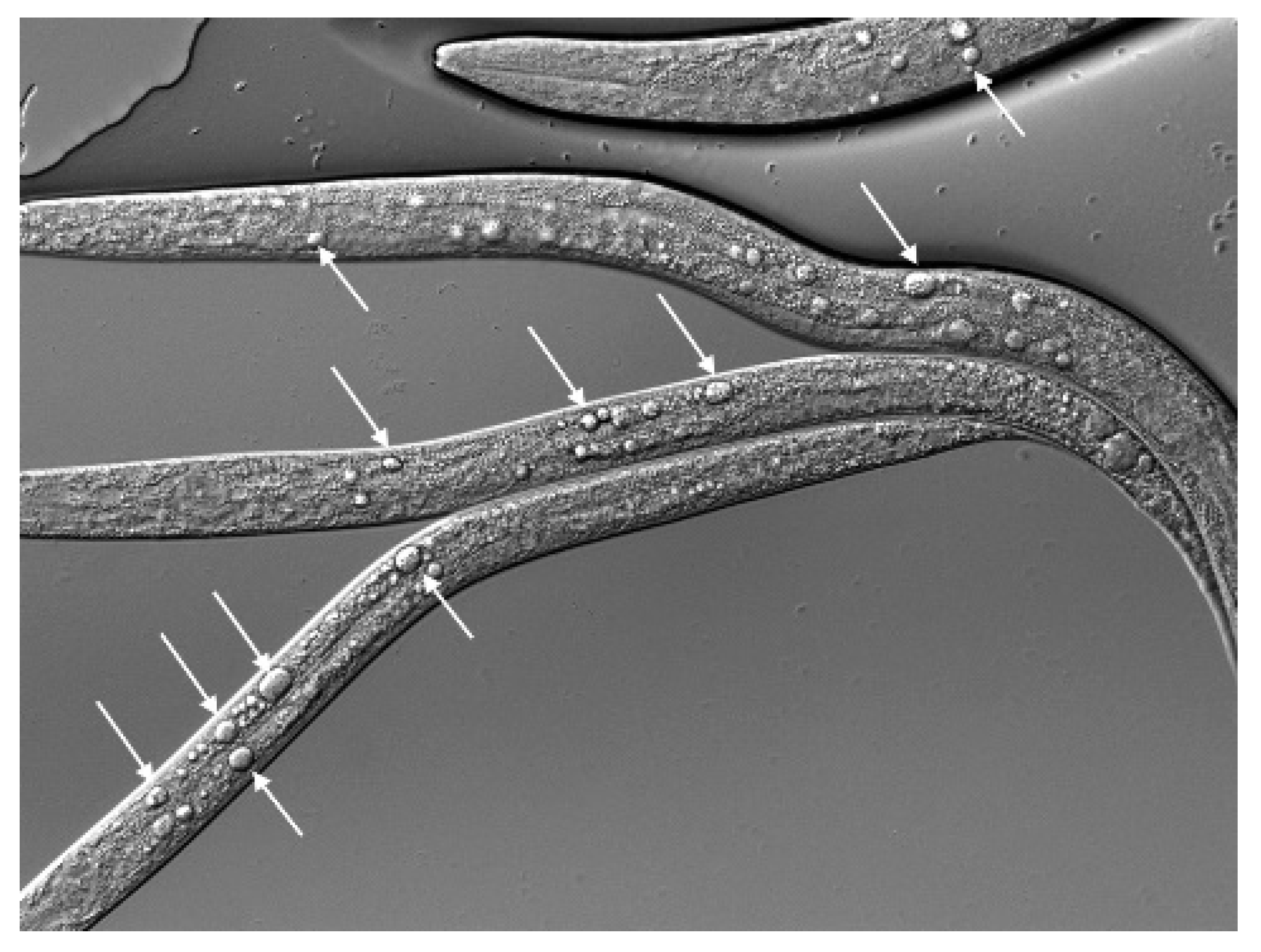
dot-1.1 and zfp-1 Mutant Worms Exhibit Gonad Migration Defects Characteristic of UNC-6/Netrin Signaling Mutants
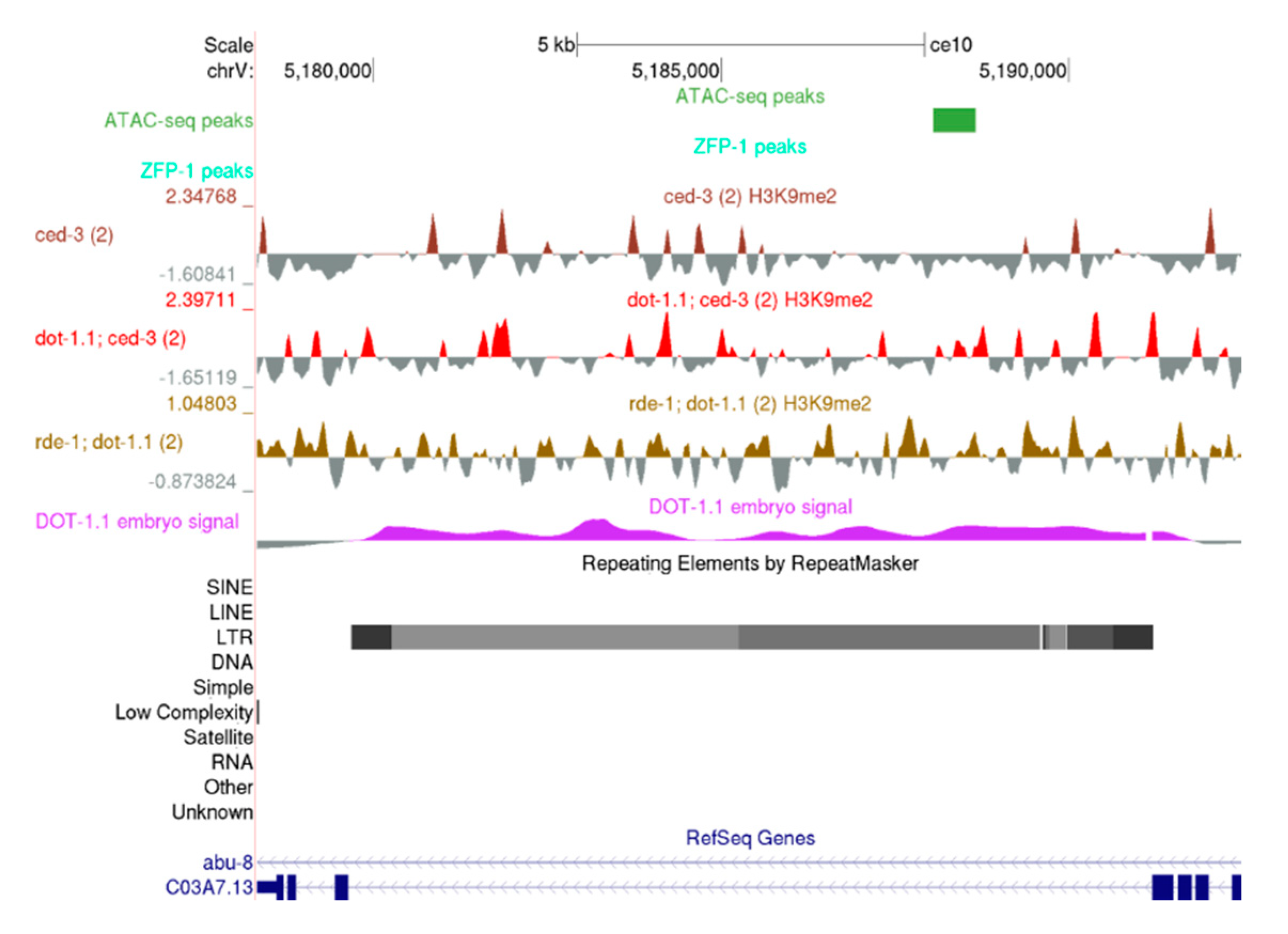
3.5. H3K9me2 Is Elevated at Repetitive Elements in dot-1.1(−) Worms
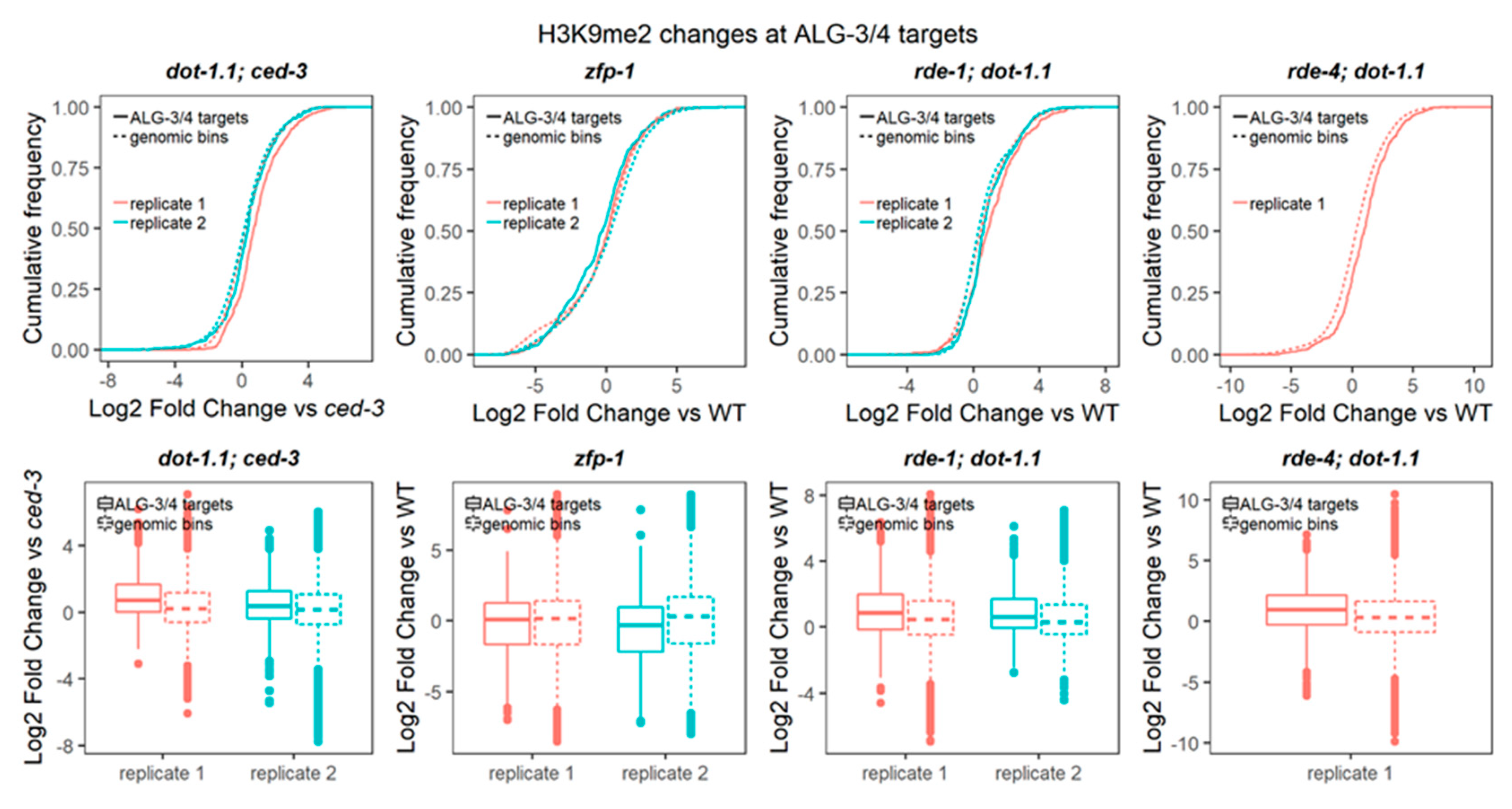
3.6. H3K9me2 Is Elevated at ALG-3/4 Target Genes in dot-1.1(−) Worms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, H.-H.; Feng, Q.; Wang, H.; Erdjument-Bromage, H.; Tempst, P.; Zhang, Y.; Struhl, K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002, 16, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Van Leeuwen, F.; Gafken, P.R.; Gottschling, D.E. Dot1p Modulates Silencing in Yeast by Methylation of the Nucleosome Core. Cell 2002, 109, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Wang, H.; Ng, H.-H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Esse, R.; Gushchanskaia, E.S.; Lord, A.; Grishok, A. DOT1L complex suppresses transcription from enhancer elements and ectopic RNAi in Caenorhabditis elegans. RNA 2019, 25, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yang, Y.; Ortega, M.M.; Copeland, J.N.; Zhang, M.; Jacob, J.B.; Fields, T.A.; Vivian, J.L.; Fields, P.E. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 2010, 116, 4483–4491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.; Su, H.; Bhat, A.; Lei, H.; Bajko, J.; Hevi, S.; Baltus, G.A.; Kadam, S.; Zhai, H.; Valdez, R.; et al. The Histone H3K79 Methyltransferase Dot1L Is Essential for Mammalian Development and Heterochromatin Structure. PLoS Genet. 2008, 4, e1000190. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Marschalek, R. Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol. 2010, 152, 141–154. [Google Scholar] [CrossRef]
- Muntean, A.G.; Hess, J.L. The Pathogenesis of Mixed-Lineage Leukemia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 283–301. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Xiao, B.; Neppl, R.L.; Kallin, E.M.; Li, J.; Chen, T.; Wang, D.-Z.; Xiao, X.; Zhang, Y. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011, 25, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; He, J.; Taranova, O.; Zhang, Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011, 21, 1370–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Xue, P.; Li, H.; Bao, Y.; Wu, L.; Chang, S.; Niu, B.; Yang, F.; Zhang, T. Histone modification mapping in human brain reveals aberrant expression of histone H3 lysine 79 dimethylation in neural tube defects. Neurobiol. Dis. 2013, 54, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Bovio, P.P.; Franz, H.; Heidrich, S.; Rauleac, T.; Kilpert, F.; Manke, T.; Vogel, T. Differential Methylation of H3K79 Reveals DOT1L Target Genes and Function in the Cerebellum In Vivo. Mol. Neurobiol. 2018, 56, 4273–4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttner, N.; Johnsen, S.A.; Kugler, S.; Vogel, T. Af9/Mllt3 interferes with Tbr1 expression through epigenetic modification of histone H3K79 during development of the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 7042–7047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, H.; Villarreal, A.; Heidrich, S.; Videm, P.; Kilpert, F.; Mestres, I.; Calegari, F.; Backofen, R.; Manke, T.; Vogel, T. DOT1L promotes progenitor proliferation and primes neuronal layer identity in the developing cerebral cortex. Nucleic Acids Res. 2018, 47, 168–183. [Google Scholar] [CrossRef]
- Chen, C.-W.; Koche, R.P.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.G.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.J.; Deshpande, A.; Sinha, A.U.; Chen, L.; Chang, J.; Cihan, A.; Fazio, M.; Chen, C.-W.; Zhu, N.; Koche, R.; et al. AF10 Regulates Progressive H3K79 Methylation and HOX Gene Expression in Diverse AML Subtypes. Cancer Cell 2014, 26, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Skucha, A.; Ebner, J.; Schmoellerl, J.; Roth, M.; Eder, T.; César-Razquin, A.; Stukalov, A.; Vittori, S.; Muhar, M.; Lu, B.; et al. MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat. Commun. 2018, 9, 1983. [Google Scholar] [CrossRef]
- Kennedy, L.M.; Grishok, A. Neuronal migration is regulated by endogenous RNAi and chromatin-binding factor ZFP-1/AF10 in Caenorhabditis elegans. Genetics 2014, 197, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.R.; Ow, M.C.; Nishiguchi, M.A.; Kim, K.; Sengupta, P.; E Hall, S. Developmental programming modulates olfactory behavior in C. elegans via endogenous RNAi pathways. ELife 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Rahe, D.P.; Hobert, O. Restriction of Cellular Plasticity of Differentiated Cells Mediated by Chromatin Modifiers, Transcription Factors and Protein Kinases. G3 Genes Genomes Genet. 2019, 9, 2287–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecere, G.; Hoersch, S.; Jensen, M.B.; Dixit, S.; Grishok, A. The ZFP-1(AF10)/DOT-1 complex opposes H2B ubiquitination to reduce Pol II transcription. Mol. Cell 2013, 50, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, M.; Herz, H.-M.; Takahashi, Y.-H.; Lin, C.; Lai, K.C.; Zhang, Y.; Washburn, M.P.; Florens, L.; Shilatifard, A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010, 24, 574–589. [Google Scholar] [CrossRef] [Green Version]
- Katan-Khaykovich, Y.; Struhl, K. Heterochromatin formation involves changes in histone modifications over multiple cell generations. EMBO J. 2005, 24, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Ahringer, J.; Gasser, S.M. Repressive Chromatin inCaenorhabditis elegans: Establishment, Composition, and Function. Genetics 2018, 208, 491–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Rechtsteiner, A.; Egelhofer, T.A.; Vielle, A.; Latorre, I.; Cheung, M.-S.; Ercan, S.; Ikegami, K.; Jensen, M.; Kolasinska-Zwierz, P.; et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2010, 21, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecere, G.; Grishok, A. A nuclear perspective on RNAi pathways in metazoans. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2013, 1839, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Guang, S. Small RNAs, RNAi and the Inheritance of Gene Silencing in Caenorhabditis elegans. J. Genet. Genom. 2013, 40, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.A.; Burkhart, K.B.; Gu, S.G.; Spracklin, G.; Kershner, A.; Fritz, H.; Kimble, J.; Fire, A.; Kennedy, S. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature 2012, 489, 447–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.Z.; Chen, E.; Gu, S.G. Complex coding of endogenous siRNA, transcriptional silencing and H3K9 methylation on native targets of germline nuclear RNAi in C. elegans. BMC Genom. 2014, 15, 1157. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.Z.; Kalinava, N.; Chen, E.; Huang, A.; Trinh, T.; Gu, S.G. A transgenerational role of the germline nuclear RNAi pathway in repressing heat stress-induced transcriptional activation in C. elegans. Epigenetics Chromatin 2016, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.Z.; Kalinava, N.; Mendoza, S.G.; Gu, S.G. The spatial and temporal dynamics of nuclear RNAi-targeted retrotransposon transcripts in Caenorhabditis elegans. Development 2018, 145, dev167346. [Google Scholar] [CrossRef] [Green Version]
- Kalinava, N.; Ni, J.Z.; Peterman, K.; Chen, E.; Gu, S.G. Decoupling the downstream effects of germline nuclear RNAi reveals that H3K9me3 is dispensable for heritable RNAi and the maintenance of endogenous siRNA-mediated transcriptional silencing in Caenorhabditis elegans. Epigenetics Chromatin 2017, 10, 6. [Google Scholar] [CrossRef]
- Kalinava, N.; Ni, J.Z.; Gajic, Z.; Kim, M.; Ushakov, H.; Gu, S.G. C. elegans Heterochromatin Factor SET-32 Plays an Essential Role in Transgenerational Establishment of Nuclear RNAi-Mediated Epigenetic Silencing. Cell Rep. 2018, 25, 2273–2284.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhouse, R.M.; Buchmann, G.; Hoe, M.; Harney, D.J.; Low, J.K.; Larance, M.; Boag, P.; Ashe, A. Chromatin Modifiers SET-25 and SET-32 Are Required for Establishment but Not Long-Term Maintenance of Transgenerational Epigenetic Inheritance. Cell Rep. 2018, 25, 2259–2272.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, O.; Edgley, M.; Strasbourger, P.; Flibotte, S.; Ewing, B.; Adair, R.; Au, V.; Chaudhry, I.; Fernando, L.; Hutter, H.; et al. The million mutation project: A new approach to genetics in Caenorhabditis elegans. Genome Res. 2013, 23, 1749–1762. [Google Scholar] [CrossRef] [Green Version]
- Avgousti, D.C.; Cecere, G.; Grishok, A. The conserved PHD1-PHD2 domain of ZFP-1/AF10 is a discrete functional module essential for viability in Caenorhabditis elegans. Mol. Cell. Biol. 2013, 33, 999–1015. [Google Scholar] [CrossRef] [Green Version]
- Mansisidor, A.; Cecere, G.; Hoersch, S.; Jensen, M.B.; Kawli, T.; Kennedy, L.M.; Chavez, V.; Tan, M.-W.; Lieb, J.D.; Grishok, A. A Conserved PHD Finger Protein and Endogenous RNAi Modulate Insulin Signaling in Caenorhabditis elegans. PLoS Genet. 2011, 7, e1002299. [Google Scholar] [CrossRef] [Green Version]
- Colaiácovo, M.P.; (Harvard Medical School, Boston, MA, USA). Personal communication, 2020.
- Tabara, H.; Yigit, E.; Siomi, H.; Mello, C.C. The dsRNA binding protein RDE-4 interacts with RDE-1, DCR-1, and a DExH-box helicase to direct RNAi in C. elegans. Cell 2002, 109, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Tabara, H.; Sarkissian, M.; Kelly, W.G.; Fleenor, J.; Grishok, A.; Timmons, L.; Fire, A.; Mello, C.C. The rde-1 Gene, RNA Interference, and Transposon Silencing in C. elegans. Cell 1999, 99, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Gaidatzis, D.; Lerch, A.; Hahne, F.; Stadler, M.B. QuasR: Quantification and annotation of short reads in R. Bioinformatics 2014, 31, 1130–1132. [Google Scholar] [CrossRef] [Green Version]
- Evans, K.J.; Huang, N.; Stempor, P.; Chesney, M.A.; Down, T.A.; Ahringer, J. Stable Caenorhabditis elegans chromatin domains separate broadly expressed and developmentally regulated genes. Proc. Natl. Acad. Sci. USA 2016, 113, E7020–E7029. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.C.; Yeo, R.W.; Buenrostro, J.D.; Greenleaf, W.J.; Kundaje, A.; Brunet, A. Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res. 2017, 27, 2096–2107. [Google Scholar] [CrossRef] [Green Version]
- Conine, C.C.; Batista, P.J.; Gu, W.; Claycomb, J.M.; Chaves, D.A.; Shirayama, M.; Mello, C.C. Argonautes ALG-3 and ALG-4 are required for spermatogenesis-specific 26G-RNAs and thermotolerant sperm in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2010, 107, 3588–3593. [Google Scholar] [CrossRef] [Green Version]
- Vasale, J.J.; Gu, W.; Thivierge, C.; Batista, P.J.; Claycomb, J.M.; Youngman, E.M.; Duchaine, T.F.; Mello, C.C.; Conte, D., Jr. Sequential rounds of RNA-dependent RNA transcription drive endogenous small-RNA biogenesis in the ERGO-1/Argonaute pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 3582–3587. [Google Scholar] [CrossRef] [Green Version]
- Rockman, M.; Kruglyak, L. Recombinational Landscape and Population Genomics of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000419. [Google Scholar] [CrossRef] [Green Version]
- McMurchy, A.N.; Stempor, P.; Gaarenstroom, T.; Wysolmerski, B.; Dong, Y.; Aussianikava, D.; Appert, A.; Huang, N.; Kolasinska-Zwierz, P.; Sapetschnig, A.; et al. A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. Elife 2017, 6, 21666. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Chung, D.; Zhang, Q.; Keleş, S. MOSAiCS-HMM: A Model-Based Approach for Detecting Regions of Histone Modifications from ChIP-Seq Data. In Statistical Analysis of Next Generation Sequencing Data; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2014; pp. 277–295. [Google Scholar]
- Robinson, M.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Chen, J.; Myers, T.R.; Hwang, B.J.; Sternberg, P.W.; Greenwald, I.; Han, M. SynMuv genes redundantly inhibit lin-3/EGF expression to prevent inappropriate vulval induction in C. elegans. Dev. Cell 2006, 10, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padeken, J.; Zeller, P.; Towbin, B.; Katic, I.; Kalck, V.; Methot, S.P.; Gasser, S.M. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev. 2019, 33, 436–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrigues, J.M.; Sidoli, S.; Garcia, B.A.; Strome, S. Defining heterochromatin in C. elegans through genome-wide analysis of the heterochromatin protein 1 homolog HPL-2. Genome Res. 2015, 25, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, P.; Padeken, J.; Van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Steiner, F.A.; Henikoff, S. Holocentromeres are dispersed point centromeres localized at transcription factor hotspots. ELife 2014, 3, 2025. [Google Scholar] [CrossRef]
- Chen, F.; Chisholm, A.; Jin, Y. Tissue-specific regulation of alternative polyadenylation represses expression of a neuronal ankyrin isoform in C. elegans epidermal development. Development 2017, 144, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Zhou, Y.; Qi, Y.B.; Khivansara, V.; Li, H.; Chun, S.Y.; Kim, J.K.; Fu, X.-D.; Jin, Y. Context-dependent modulation of Pol II CTD phosphatase SSUP-72 regulates alternative polyadenylation in neuronal development. Genes Dev. 2015, 29, 2377–2390. [Google Scholar] [CrossRef] [Green Version]
- Colavita, A.; Culotti, J.G. Suppressors of Ectopic UNC-5 Growth Cone Steering Identify Eight Genes Involved in Axon Guidance inCaenorhabditis elegans. Dev. Biol. 1998, 194, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Hedgecock, E.M.; Culotti, J.G.; Thomson, J.; Perkins, L.A. Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev. Biol. 1985, 111, 158–170. [Google Scholar] [CrossRef]
- Meng, L.; Chen, C.H.; Yan, D. Regulation of Gap Junction Dynamics by UNC-44/ankyrin and UNC-33/CRMP through VAB-8 in C. elegans Neurons. PLoS Genet. 2016, 12, e1005948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, A.J.; Boontrakulpoontawee, P.; Rebeiz, N.; Domanus, M.; Otsuka, D.; Velamparampil, N.; Chan, S.; Wyngaerde, M.V.; Campagna, S.; Cox, A. Novel UNC-44 AO13 ankyrin is required for axonal guidance in C. elegans, contains six highly repetitive STEP blocks separated by seven potential transmembrane domains, and is localized to neuronal processes and the periphery of neural cell bodies. J. Neurobiol. 2002, 50, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.J.; Franco, R.; Yang, B.; Shim, K.H.; Tang, L.Z.; Zhang, Y.Y.; Boontrakulpoontawee, P.; Jeyaprakash, A.; Hedgecock, E.; I Wheaton, V. An ankyrin-related gene (unc-44) is necessary for proper axonal guidance in Caenorhabditis elegans. J. Cell Biol. 1995, 129, 1081–1092. [Google Scholar] [CrossRef] [Green Version]
- A Zallen, J.; A Kirch, S.; I Bargmann, C. Genes required for axon pathfinding and extension in the C. elegans nerve ring. Development 1999, 126, 3679–3692. [Google Scholar]
- Zhou, S.; Opperman, K.; Wang, X.; Chen, L. unc-44 Ankyrin and stn-2 gamma-syntrophin regulate sax-7 L1CAM function in maintaining neuronal positioning in Caenorhabditis elegans. Genetics 2008, 180, 1429–1443. [Google Scholar] [CrossRef] [Green Version]
- Brundage, L.; Avery, L.; Katz, A.; Kim, U.J.; Mendel, J.E.; Sternberg, P.W.; Simon, M.I. Mutations in a C. elegans Gqalpha gene disrupt movement, egg laying, and viability. Neuron 1996, 16, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Lackner, M.R.; Nurrish, S.J.; Kaplan, J.M. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 1999, 24, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Tanis, J.E.; Moresco, J.J.; Lindquist, R.A.; Koelle, M.R. Regulation of serotonin biosynthesis by the G proteins Galphao and Galphaq controls serotonin signaling in Caenorhabditis elegans. Genetics 2008, 178, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Shyn, S.I.; Kerr, R.; Schafer, W.R. Serotonin and Go Modulate Functional States of Neurons and Muscles Controlling C. elegans Egg-Laying Behavior. Curr. Biol. 2003, 13, 1910–1915. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Kunitomo, H.; Tomioka, M.; Ohno, H.; Okochi, Y.; Mori, I.; Iino, Y. Reversal of Salt Preference Is Directed by the Insulin/PI3K and Gq/PKC Signaling in Caenorhabditis elegans. Genetics 2010, 186, 1309–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastuhov, S.I.; Fujiki, K.; Nix, P.; Kanao, S.; Bastiani, M.; Matsumoto, K.; Hisamoto, N. Endocannabinoid-Goalpha signalling inhibits axon regeneration in Caenorhabditis elegans by antagonizing Gqalpha-PKC-JNK signalling. Nat. Commun. 2012, 3, 1136. [Google Scholar] [CrossRef] [Green Version]
- Aboobaker, A.A.; Blaxter, M.L. Hox Gene Loss during Dynamic Evolution of the Nematode Cluster. Curr. Biol. 2003, 13, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Dudley, N.R.; Labbé, J.-C.; Goldstein, B. Using RNA interference to identify genes required for RNA interference. Proc. Natl. Acad. Sci. USA 2002, 99, 4191–4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.R.; Li, W.; et al. AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, M.; Kim, E.B.; Han, M. Diverse Chromatin Remodeling Genes Antagonize the Rb-Involved SynMuv Pathways in C. elegans. PLoS Genet. 2006, 2, e74. [Google Scholar] [CrossRef] [Green Version]
- Cram, E.J.; Shang, H.; Schwarzbauer, J.E. A systematic RNA interference screen reveals a cell migration gene network in C. elegans. J. Cell Sci. 2006, 119, 4811–4818. [Google Scholar] [CrossRef] [Green Version]
- Hedgecock, E.M.; Culotti, J.G.; Hall, D.H. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron 1990, 4, 61–85. [Google Scholar] [CrossRef]
- Sun, K.L.W.; Correia, J.P.; Kennedy, T.E. Netrins: Versatile extracellular cues with diverse functions. Development 2011, 138, 2153–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadsworth, W.G.; Hedgecock, E.M. Hierarchical guidance cues in the developing nervous system ofC. elegans. BioEssays 1996, 18, 355–362. [Google Scholar] [CrossRef]
- Grishok, A. Biology and Mechanisms of Short RNAs in Caenorhabditis elegans. Adv. Genet. 2013, 83, 1–69. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Manoharan, A.P.; Harkins, T.T.; Bouffard, P.; Fitzpatrick, C.; Chu, D.S.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, J.K. 26G endo-siRNAs regulate spermatogenic and zygotic gene expression in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2009, 106, 18674–18679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloss, T.A.; Witze, E.S.; Rothman, J.H. Suppression of CED-3-independent apoptosis by mitochondrial βNAC in Caenorhabditis elegans. Nature 2003, 424, 1066–1071. [Google Scholar] [CrossRef]
- Godfrey, L.; Crump, N.T.; O’Byrne, S.; Lau, I.J.; Rice, S.; Harman, J.R.; Jackson, T.; Elliott, N.; Buck, G.; Connor, C.; et al. H3K79me2/3 controls enhancer-promoter interactions and activation of the pan-cancer stem cell marker PROM1/CD133 in MLL-AF4 leukemia cells. Leukemia 2020, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, L.; Crump, N.T.; Thorne, R.; Lau, I.-J.; Repapi, E.; Dimou, D.; Smith, A.L.; Harman, J.R.; Telenius, J.M.; Oudelaar, A.M.; et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Zhao, Z.; He, F.; Du, Z. Multivariable regulation of gene expression plasticity in metazoans. Open Biol. 2019, 9, 190150. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.I.S. RNAi-dependent formation of heterochromatin and its diverse functions. Curr. Opin. Genet. Dev. 2010, 20, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.B.; Sung, S. Vernalization-Mediated Epigenetic Silencing by a Long Intronic Noncoding RNA. Science 2010, 331, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Świezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar] [CrossRef]
- Long, Y.; Bolanos, B.; Gong, L.; Liu, W.; Goodrich, K.J.; Yang, X.; Chen, S.; Gooding, A.R.; A Maegley, K.; Gajiwala, K.S.; et al. Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. ELife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Dutta, B.; Hee, Y.T.; Chng, W.-J. Towards understanding of PRC2 binding to RNA. RNA Biol. 2019, 16, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gushchanskaia, E.S.; Esse, R.; Ma, Q.; Lau, N.; Grishok, A. Interplay between small RNA pathways shapes chromatin landscapes in C. elegans. Nucleic Acids Res. 2019, 47, 5603–5616. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | % with Phenotype | Total Gonads 1 |
|---|---|---|
| Wild type (N2) | 0 | 100 |
| dot-1.1(gk520244) | 44 | 166 |
| zfp-1(gk960739) | 5 | 232 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esse, R.; Grishok, A. Caenorhabditis elegans Deficient in DOT-1.1 Exhibit Increases in H3K9me2 at Enhancer and Certain RNAi-Regulated Regions. Cells 2020, 9, 1846. https://doi.org/10.3390/cells9081846
Esse R, Grishok A. Caenorhabditis elegans Deficient in DOT-1.1 Exhibit Increases in H3K9me2 at Enhancer and Certain RNAi-Regulated Regions. Cells. 2020; 9(8):1846. https://doi.org/10.3390/cells9081846
Chicago/Turabian StyleEsse, Ruben, and Alla Grishok. 2020. "Caenorhabditis elegans Deficient in DOT-1.1 Exhibit Increases in H3K9me2 at Enhancer and Certain RNAi-Regulated Regions" Cells 9, no. 8: 1846. https://doi.org/10.3390/cells9081846
APA StyleEsse, R., & Grishok, A. (2020). Caenorhabditis elegans Deficient in DOT-1.1 Exhibit Increases in H3K9me2 at Enhancer and Certain RNAi-Regulated Regions. Cells, 9(8), 1846. https://doi.org/10.3390/cells9081846