Hepatitis B Virus Exploits ERGIC-53 in Conjunction with COPII to Exit Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression Constructs
2.2. SiRNAs, Cell Culture and Transfection
2.3. Antibodies
2.4. Quantitative Reverse Transcription PCR (qRT-PCR) Analysis
2.5. Subviral Particle Analysis
2.6. Viral Particle Analysis
2.7. Endoglycosidase H (EndoH) Treatment
2.8. Coimmunoprecipitation (CO-IP) Assay
2.9. Fluorescence Microscopy
2.10. Statistics
3. Results
3.1. HBV L Resides within Markedly Crescent-Shaped ER-Associated Compartments
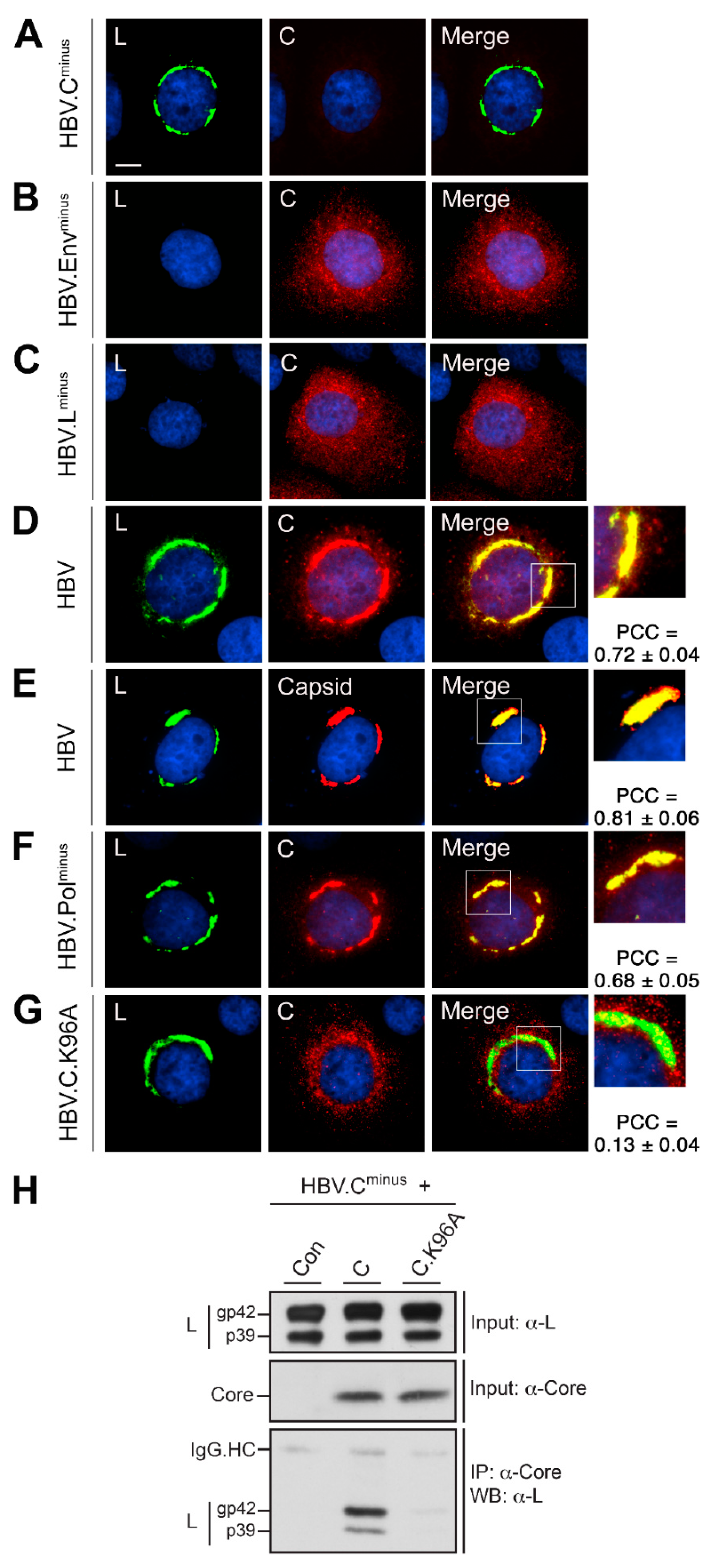
3.2. HBV L Recruits Core/Capsid to the Crescent-Shaped ER-Associated Compartments
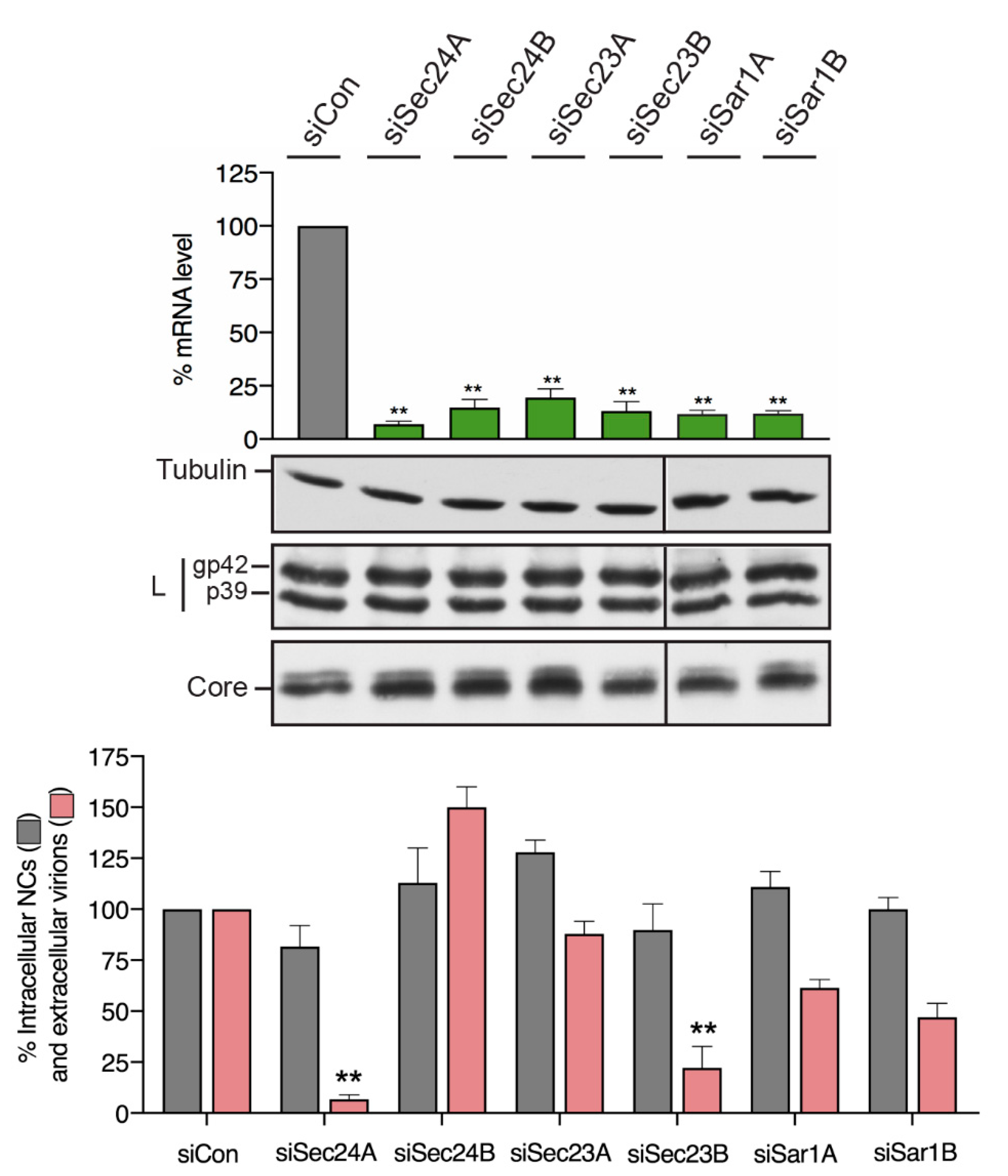
3.3. HBV Particle Egress Requires the COPII components Sec24A, Sec23B and Sar1
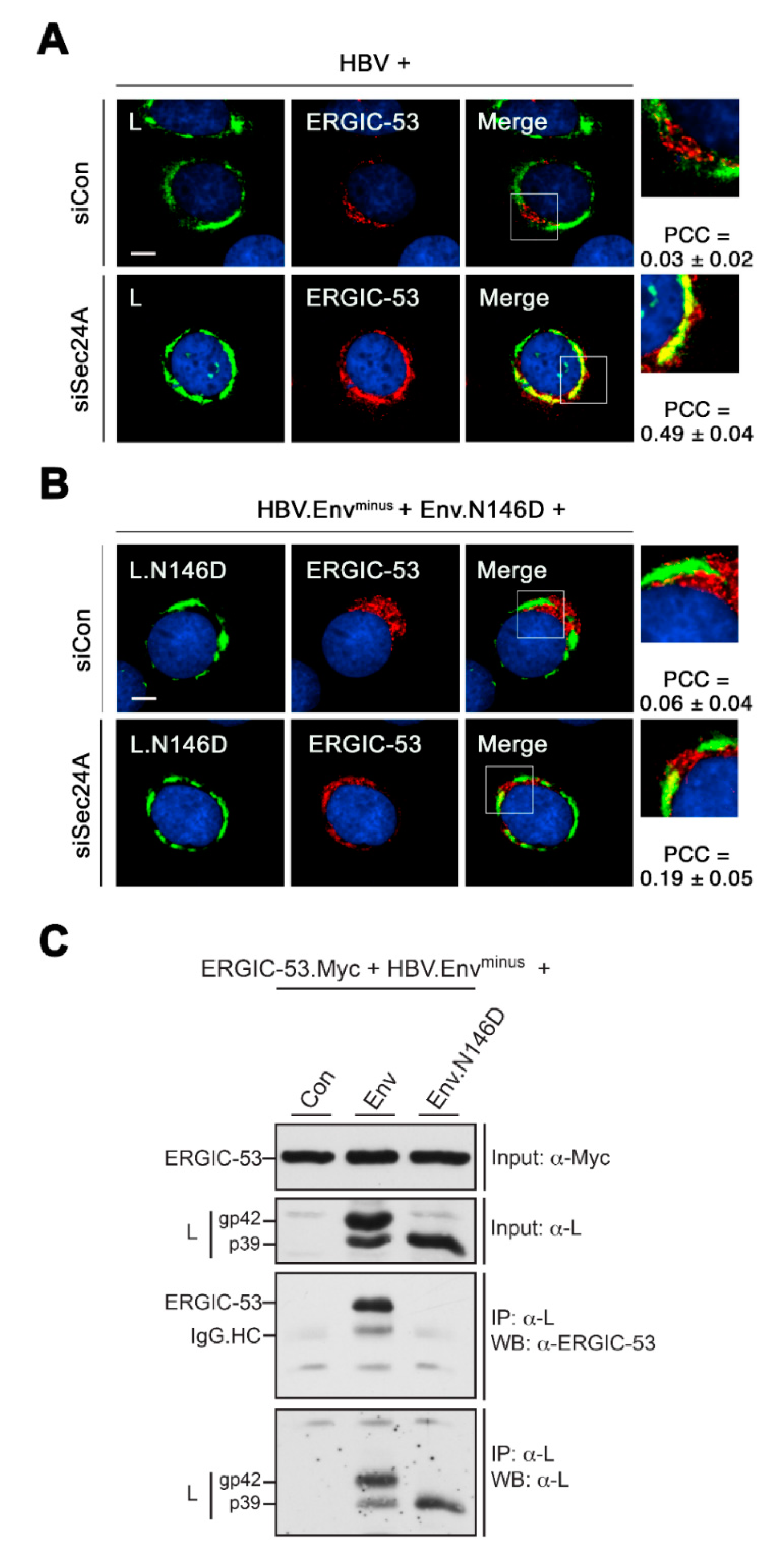
3.4. HBV L Colocalizes with ERGIC-53 upon ER Export Block
3.5. HBV Particle Egress Requires ERGIC-53, while HBV Spherical Subviral Particle Egress Does Not
3.6. Blocking N146-linked Glycosylation Inhibits HBV Egress without Affecting Envelope/Core Interactions
3.7. HBV L Interacts with ERGIC-53 in a N146-Glycan-Dependent Manner
3.8. HBV Envelopment of Nucleocapsids Occurs prior to the Actions of ERGIC-53 and Sec24A
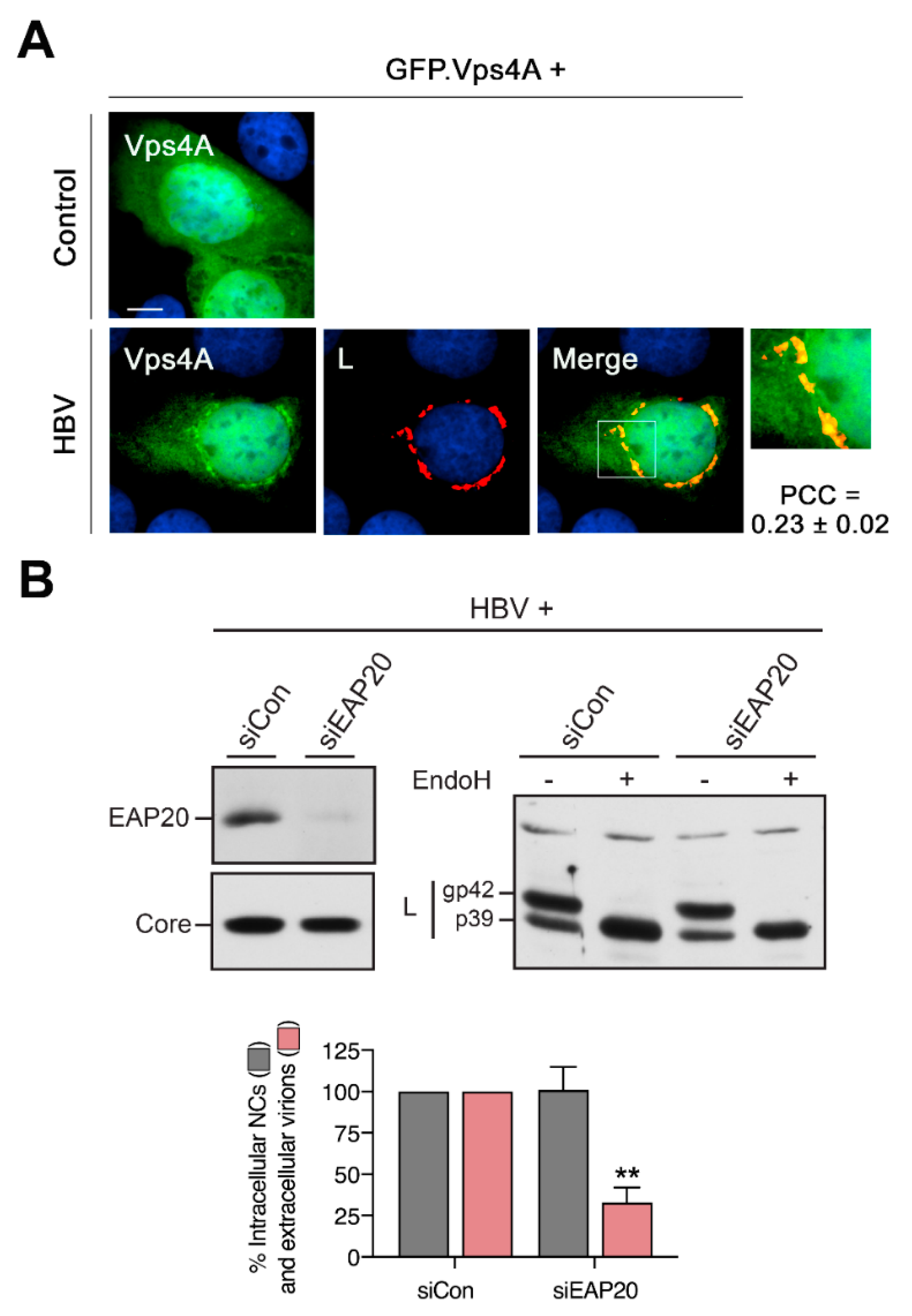
3.9. HBV Recruits the ESCRT-specific Vps4 ATPase to the Crescent-Shaped ER-Associated Compartments
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Mitra, B.; Thapa, R.J.; Guo, H.; Block, T.M. Host functions used by hepatitis B virus to complete its life cycle: Implications for developing host-targeting agents to treat chronic hepatitis B. Antivir. Res. 2018, 158, 185–198. [Google Scholar] [CrossRef]
- Hu, J.; Seeger, C. Hepadnavirus Genome Replication and Persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Blondot, M.-L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef]
- Li, W. The Hepatitis B Virus Receptor. Annu. Rev. Cell Dev. Boil. 2015, 31, 125–147. [Google Scholar] [CrossRef]
- Bruss, V.; Ganem, D. The role of envelope proteins in hepatitis B virus assembly. Proc. Natl. Acad. Sci. USA 1991, 88, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Bruss, V. A short linear sequence in the pre-S domain of the large hepatitis B virus envelope protein required for virion formation. J. Virol. 1997, 71, 9350–9357. [Google Scholar] [CrossRef] [Green Version]
- Prange, R.; Streeck, R. Novel transmembrane topology of the hepatitis B virus envelope proteins. EMBO J. 1995, 14, 247–256. [Google Scholar] [CrossRef]
- Bi, X.; Tong, S. Impact of immune escape mutations and N-linked glycosylation on the secretion of hepatitis B virus virions and subviral particles: Role of the small envelope protein. Virol. 2018, 518, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Qin, Y.; Guarnieri, M.; Garcia, T.; Kwei, K.; Mizokami, M.; Zhang, J.; Li, J.; Wands, J.R.; Tong, S. Impairment of Hepatitis B Virus Virion Secretion by Single-Amino-Acid Substitutions in the Small Envelope Protein and Rescue by a Novel Glycosylation Site. J. Virol. 2010, 84, 12850–12861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julithe, R.; Abou-Jaoudé, G.; Sureau, C. Modification of the Hepatitis B Virus Envelope Protein Glycosylation Pattern Interferes with Secretion of Viral Particles, Infectivity, and Susceptibility to Neutralizing Antibodies. J. Virol. 2014, 88, 9049–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Mehta, A.; Dwek, R.; Butters, T.; Block, T.M. Evidence That N-Linked Glycosylation Is Necessary for Hepatitis B Virus Secretion. Virol. 1995, 213, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Yang, F. Post-translational Modification Control of HBV Biological Processes. Front. Microbiol. 2018, 9, 2661. [Google Scholar] [CrossRef]
- Dobrica, M.-O.; Lazar, C.; Branza-Nichita, N. N-Glycosylation and N-Glycan Processing in HBV Biology and Pathogenesis. Cells 2020, 9, 9. [Google Scholar] [CrossRef]
- Hassemer, M.; Finkernagel, M.; Peiffer, K.-H.; Glebe, D.; Akhras, S.; Reuter, A.; Scheiblauer, H.; Sommer, L.; Chudy, M.; Nübling, C.M.; et al. Comparative characterization of hepatitis B virus surface antigen derived from different hepatitis B virus genotypes. Virol. 2017, 502, 1–12. [Google Scholar] [CrossRef]
- Le Seyec, J.; Chouteau, P.; Cannie, I.; Guguen-Guillouzo, C.; Gripon, P. Infection Process of the Hepatitis B Virus Depends on the Presence of a Defined Sequence in the Pre-S1 Domain. J. Virol. 1999, 73, 2052–2057. [Google Scholar] [CrossRef] [Green Version]
- Tolle, T.K.; Glebe, D.; Linder, M.; Linder, D.; Schmitt, S.; Geyer, R.; Gerlich, W.H. Structure and Glycosylation Patterns of Surface Proteins from Woodchuck Hepatitis Virus. J. Virol. 1998, 72, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Lambert, C.; Döring, T.; Prange, R. Hepatitis B Virus Maturation Is Sensitive to Functional Inhibition of ESCRT-III, Vps4, and γ2-Adaptin. J. Virol. 2007, 81, 9050–9060. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieler, J.T.; Prange, R. Involvement of ESCRT-II in Hepatitis B Virus Morphogenesis. PLoS ONE 2014, 9, e91279. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2015, 90, 3330–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.-F.; Tsai, M.-L.; Huang, J.-Y.; Chang, Y.-S.; Shih, C. The Dual Role of an ESCRT-0 Component HGS in HBV Transcription and Naked Capsid Secretion. PLOS Pathog. 2015, 11, e1005123. [Google Scholar] [CrossRef] [Green Version]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Boil. 2019, 21, 25–42. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Mohebbi, A.; Lorestani, N.; Tahamtan, A.; Kargar, N.L.; Tabarraei, A. An Overview of Hepatitis B Virus Surface Antigen Secretion Inhibitors. Front. Microbiol. 2018, 9, 662. [Google Scholar] [CrossRef] [Green Version]
- Huovila, A.P.; Eder, A.M.; Fuller, S.D. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J. Cell Boil. 1992, 118, 1305–1320. [Google Scholar] [CrossRef]
- Zeyen, L.; Döring, T.; Stieler, J.T.; Prange, R. Hepatitis B subviral envelope particles use the COPII machinery for intracellular transport via selective exploitation of Sec24A and Sec23B. Cell. Microbiol. 2020, 22, e13181. [Google Scholar] [CrossRef] [Green Version]
- Khoriaty, R.; Vasievich, M.P.; Ginsburg, D. The COPII pathway and hematologic disease. Blood 2012, 120, 31–38. [Google Scholar] [CrossRef]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Radziwill, G.; Tucker, W.; Schaller, H. Mutational analysis of the hepatitis B virus P gene product: Domain structure and RNase H activity. J. Virol. 1990, 64, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffler-Mary, H.; Dumortier, J.; Klentsch-Zimmer, C.; Prange, R. Hepatitis B Virus Assembly Is Sensitive to Changes in the Cytosolic S Loop of the Envelope Proteins. Virology 2000, 270, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, M.; Mann, S.; Lambert, C.; Döring, T.; Thomé, N.; Prange, R. γ2-Adaptin, a Novel Ubiquitin-interacting Adaptor, and Nedd4 Ubiquitin Ligase Control Hepatitis B Virus Maturation. J. Boil. Chem. 2006, 281, 29297–29308. [Google Scholar] [CrossRef] [Green Version]
- Bardens, A.; Döring, T.; Stieler, J.T.; Prange, R. Alix regulates egress of hepatitis B virus naked capsid particles in an ESCRT-independent manner. Cell. Microbiol. 2010, 13, 602–619. [Google Scholar] [CrossRef]
- Mangold, C.M.; Streeck, R.E. Mutational analysis of the cysteine residues in the hepatitis B virus small envelope protein. J. Virol. 1993, 67, 4588–4597. [Google Scholar] [CrossRef] [Green Version]
- Nyfeler, B.; Reiterer, V.; Wendeler, M.W.; Stefan, E.; Zhang, B.; Michnick, S.W.; Hauri, H.-P. Identification of ERGIC-53 as an intracellular transport receptor of α1-antitrypsin. J. Cell Boil. 2008, 180, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Ronzoni, R.; Anelli, T.; Brunati, M.; Cortini, M.; Fagioli, C.; Sitia, R. Pathogenesis of ER Storage Disorders: Modulating Russell Body Biogenesis by Altering Proximal and Distal Quality Control. Traffic 2010, 11, 947–957. [Google Scholar] [CrossRef]
- Klaus, J.P.; Eisenhauer, P.; Russo, J.; Mason, A.B.; Do, D.C.; King, B.; Taatjes, D.; Cornillez-Ty, C.; Boyson, J.E.; Thali, M.; et al. The intracellular cargo receptor ERGIC-53 is required for the production of infectious arenavirus, coronavirus, and filovirus particles. Cell Host Microbe 2013, 14, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Bartusch, C.; Döring, T.; Prange, R. Rab33B Controls Hepatitis B Virus Assembly by Regulating Core Membrane Association and Nucleocapsid Processing. Viruses 2017, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Doring, T.; Zeyen, L.; Bartusch, C.; Prange, R. Hepatitis B Virus Subverts the Autophagy Elongation Complex Atg5-12/16L1 and Does Not Require Atg8/LC3 Lipidation for Viral Maturation. J. Virol. 2018, 92, JVI.01513-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenzeller-Herzog, C.; Andersson, H.; Kappeler, F.; Hauri, H.-P. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat. Cell Biol. 1999, 1, 330–334. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Junker-Niepmann, M.; Schaller, H. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 1990, 64, 5324–5332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponsel, D.; Bruss, V. Mapping of Amino Acid Side Chains on the Surface of Hepatitis B Virus Capsids Required for Envelopment and Virion Formation. J. Virol. 2003, 77, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patient, R.; Hourioux, C.; Roingeard, P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 2009, 11, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Wendeler, M.W.; Paccaud, J.-P.; Hauri, H.-P. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 2007, 8, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Khoriaty, R.; Hesketh, G.G.; Bernard, A.; Weyand, A.C.; Mellacheruvu, D.; Zhu, G.; Hoenerhoff, M.J.; McGee, B.; Everett, L.; Adams, E.J.; et al. Functions of the COPII gene paralogs SEC23A and SEC23B are interchangeable in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E7748–E7757. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, G.; Liu, Y.; Liu, S.; Aridor, M.; Huang, Y.; Hu, Y.; Wang, L.; Li, S.; Xiong, H.; et al. Small GTPases SAR1A and SAR1B regulate the trafficking of the cardiac sodium channel Nav1.5. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3672–3684. [Google Scholar] [CrossRef]
- Klumperman, J.; Schweizer, A.; Clausen, H.; Tang, B.L.; Hong, W.; Oorschot, V.; Hauri, H.P. The recycling pathway of protein ERGIC-53 and dynamics of the ER-Golgi intermediate compartment. J. Cell Sci. 1998, 111, 3411–3425. [Google Scholar]
- Zhang, Y.C.; Zhou, Y.; Yang, C.Z.; Xiong, D.S. A review of ERGIC-53: Its structure, functions, regulation and relations with diseases. Histol. Histopathol. 2009, 24, 1193–1204. [Google Scholar]
- Nebenführ, A.; Ritzenthaler, C.; Robinson, D.G. Brefeldin A: Deciphering an Enigmatic Inhibitor of Secretion. Plant. Physiol. 2002, 130, 1102–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, T.; Li, J.; Sureau, C.; Ito, K.; Qin, Y.; Wands, J.; Tong, S. Drastic Reduction in the Production of Subviral Particles Does Not Impair Hepatitis B Virus Virion Secretion. J. Virol. 2009, 83, 11152–11165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenzeller-Herzog, C.; Roche, A.-C.; Nufer, O.; Hauri, H.-P. pH-induced Conversion of the Transport Lectin ERGIC-53 Triggers Glycoprotein Release. J. Boil. Chem. 2004, 279, 12943–12950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Goldberg, J. The Transport Signal on Sec22 for Packaging into COPII-Coated Vesicles Is a Conformational Epitope. Mol. Cell 2007, 26, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.C.; Seligsohn, U.; Zivelin, A.; Terry, V.H.; Hertel, C.E.; Wheatley, M.; Moussalli, M.J.; Hauri, H.-P.; Ciavarella, N.; Kaufman, R.J.; et al. Mutations in the ER–Golgi Intermediate Compartment Protein ERGIC-53 Cause Combined Deficiency of Coagulation Factors V and VIII. Cell 1998, 93, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Nufer, O.; Kappeler, F.; Guldbrandsen, S.; Hauri, H.-P. ER export of ERGIC-53 is controlled by cooperation of targeting determinants in all three of its domains. J. Cell Sci. 2003, 116, 4429–4440. [Google Scholar] [CrossRef] [Green Version]
- Duellman, T.; Burnett, J.; Shin, A.; Yang, J. LMAN1 (ERGIC-53) is a potential carrier protein for matrix metalloproteinase-9 glycoprotein secretion. Biochem. Biophys. Res. Commun. 2015, 464, 685–691. [Google Scholar] [CrossRef] [Green Version]
- Neerman-Arbez, M.; Johnson, K.; Morris, M.; McVey, J.; Peyvandi, F.; Nichols, W.; Ginsburg, D.; Rossier, C.; Antonarakis, S.; Tuddenham, E. Molecular Analysis of the ERGIC-53 Gene in 35 Families with Combined Factor V-Factor VIII Deficiency. Blood 1999, 93, 2253–2260. [Google Scholar] [CrossRef]
- Vollenweider, F.; Kappeler, F.; Itin, C.; Hauri, H.-P. Mistargeting of the Lectin ERGIC-53 to the Endoplasmic Reticulum of HeLa Cells Impairs the Secretion of a Lysosomal Enzyme. J. Cell Boil. 1998, 142, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Appenzeller-Herzog, C.; Nyfeler, B.; Burkhard, P.; Santamarí, Í.; López-Otín, C.; Hauri, H.-P. Carbohydrate- and Conformation-dependent Cargo Capture for ER-ExitD. Mol. Boil. Cell 2005, 16, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hojo, S.; Matsumoto, N.; Yamamoto, K. Regulation of Mac-2BP secretion is mediated by its N-glycan binding to ERGIC-53. Glycobiol. 2013, 23, 904–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Page, R.C.; Das, V.; Nix, J.C.; Wigren, E.; Misra, S.; Zhang, B. Structural Characterization of Carbohydrate Binding by LMAN1 Protein Provides New Insight into the Endoplasmic Reticulum Export of Factors V (FV) and VIII (FVIII). J. Boil. Chem. 2013, 288, 20499–20509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruss, V.; Lu, X.; Thomssen, R.; Gerlich, W. Post-translational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J. 1994, 13, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Herrscher, C.; Patient, R.; Eymieux, S.; Moreau, A.; Burlaud-Gaillard, J.; Seigneuret, F.; De Rocquigny, H.; Roingeard, P.; Hourioux, C. Direct interaction between the hepatitis B virus core and envelope proteins analyzed in a cellular context. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Luckenbaugh, L.; Liu, K.; Bruss, V.; Sureau, C.; Hu, J. Common and Distinct Capsid and Surface Protein Requirements for Secretion of Complete and Genome-Free Hepatitis B Virions. J. Virol. 2018, 92, JVI.00272–18. [Google Scholar] [CrossRef] [Green Version]
- Geva, Y.; Schuldiner, M. The Back and Forth of Cargo Exit from the Endoplasmic Reticulum. Curr. Boil. 2014, 24, R130–R136. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhao, X.; Cai, D.; Liu, Y.; Block, T.M.; Guo, J.-T.; Guo, H. The Interferon-Inducible Protein Tetherin Inhibits Hepatitis B Virus Virion Secretion. J. Virol. 2015, 89, 9200–9212. [Google Scholar] [CrossRef] [Green Version]
- Inoue, J.; Krueger, E.W.; Chen, J.; Cao, H.; Ninomiya, M.; McNiven, M.A. HBV secretion is regulated through the activation of endocytic and autophagic compartments mediated by Rab7 stimulation. J. Cell Sci. 2015, 128, 1696–1706. [Google Scholar] [CrossRef] [Green Version]
- Lazar, C.; Macovei, A.; Petrescu, S.; Branza-Nichita, N. Activation of ERAD Pathway by Human Hepatitis B Virus Modulates Viral and Subviral Particle Production. PLoS ONE 2012, 7, e34169. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, C.; Wang, X.; Kemper, T.; Squire, A.; Gunzer, M.; Zhang, J.; Chen, X.; Lu, M. Hepatitis B virus is degraded by autophagosome-lysosome fusion mediated by Rab7 and related components. Protein Cell 2019, 10, 60–66. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeyen, L.; Döring, T.; Prange, R. Hepatitis B Virus Exploits ERGIC-53 in Conjunction with COPII to Exit Cells. Cells 2020, 9, 1889. https://doi.org/10.3390/cells9081889
Zeyen L, Döring T, Prange R. Hepatitis B Virus Exploits ERGIC-53 in Conjunction with COPII to Exit Cells. Cells. 2020; 9(8):1889. https://doi.org/10.3390/cells9081889
Chicago/Turabian StyleZeyen, Lisa, Tatjana Döring, and Reinhild Prange. 2020. "Hepatitis B Virus Exploits ERGIC-53 in Conjunction with COPII to Exit Cells" Cells 9, no. 8: 1889. https://doi.org/10.3390/cells9081889
APA StyleZeyen, L., Döring, T., & Prange, R. (2020). Hepatitis B Virus Exploits ERGIC-53 in Conjunction with COPII to Exit Cells. Cells, 9(8), 1889. https://doi.org/10.3390/cells9081889