Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review

 ,
,  , and
, and 
Abstract
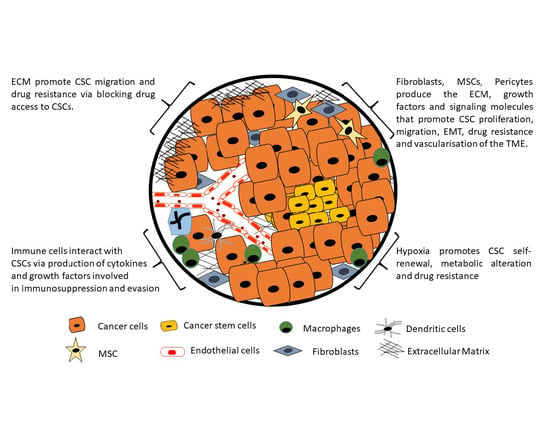
:
1. Introduction
2. Properties of Cancer Stem Cells
2.1. Cancer Stem Cell Markers and Therapy Resistance
2.2. Cancer Stem Cells and Angiogenesis
2.3. Cancer Stem Cells and Epithelial to Mesenchymal Transition
2.4. Cancer Stem Cells and Metabolic Activity
2.5. Cancer Stem Cells and Epigenetic Reprogramming
3. Targeting Cancer Stem Cells in Tumor Microenvironment
3.1. Targeting Cancer Stem Cell Signaling
3.2. Targeting Cancer-Stem-Cell-Associated Tumor Angiogenesis and Metastasis
3.3. Targeting the Immune System to Eradicate Cancer Stem Cells
3.4. Targeting Epigenetic Modifications in Cancer Stem Cells
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ALDH1 | aldehyde dehydrogenase 1 |
| ATRA | All-trans retinoic acid |
| BMI1 | B-cell-specific Moloney murine leukemia virus integration site 1 |
| BMP-4 | Bone morphogenic protein 4 |
| CAR T | Chimeric antigen receptor |
| CCA | Cholangiocarcinoma |
| CK | cytokeratin |
| CNS | central nervous system |
| CRC | Colorectal Cancer |
| CSCs | Cancer stem cells |
| CTL | Cytotoxic T lymphocyte |
| DCLK1 | Double cortin-like kinase 1; |
| EMA | epithelial membrane antigen; |
| EMT | Epithelial to mesenchymal transition |
| EpCAM | epithelial cell adhesion molecule; |
| GCSF | Granulocyte colony stimulation factor |
| H3K27me3 | Histone 3 lysine 27 |
| HDACs | Histone deacetylases |
| HDMs | Histone demethylase |
| HMTs | Histone methyltransferase |
| HNSCC | head and neck squamous cell carcinoma |
| K17 | Keratin 17 |
| Lgr5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| MDR1 | Multidrug resistance 1 |
| MGMT | O6-methylguanine DNA methyltransferase |
| MMPs | Matrix metalloproteases |
| PD-1 | Programmed cell death 1 |
| PD-L1 | PD-1 ligand |
| PSA | prostate-specific antigen |
| TME | Tumor microenvironment |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TMZ | Temozolomide |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2015, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef] [Green Version]
- Al Faraj, A.; Shaik, A.S.; Al Sayed, B.; Halwani, R.; Al Jammaz, I. Specific targeting and noninvasive imaging of breast cancer stem cells using single-walled carbon nanotubes as novel multimodality nanoprobes. Nanomedicine 2016, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, H.; Hao, X. The self-renewal signaling pathways utilized by gastric cancer stem cells. Tumor Boil. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Humphries, H.N.; Wickremesekera, S.K.; Marsh, R.W.; Brasch, H.D.; Mehrotra, S.; Tan, S.T.; Itinteang, T. Characterization of Cancer Stem Cells in Colon Adenocarcinoma Metastasis to the Liver. Front. Surg. 2018, 4, 76. [Google Scholar] [CrossRef]
- Geng, S.-Q.; Alexandrou, A.T.; Li, J.J. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014, 349, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A Human Perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Jiang, X.; Jorgensen, H.G.; Graham, S.; Alcorn, M.J.; Laird, C.; Eaves, A.C.; Eaves, C.J. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood 2001, 97, 720–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Ther. 2016, 158, 71–90. [Google Scholar] [CrossRef]
- Maugeri-Saccà, M.; Bartucci, M.; De Maria, R. DNA Damage Repair Pathways in Cancer Stem Cells. Mol. Cancer Ther. 2012, 11, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Maugeri-Saccà, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [Green Version]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Boil. 2019, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.-H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2007, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Agliano, A.; Calvo, A.; Box, C. The challenge of targeting cancer stem cells to halt metastasis. Semin. Cancer Boil. 2017, 44, 25–42. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Putten, M. The use of genetically humanized animal models for personalized medicine approaches. Dis. Model. Mech. 2019, 13, dmm041673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, Y.; Beatty, C.; Biradar, S.; Castronova, I.; Ho, S.; Melody, K.; Bility, M.T. Moving beyond the mousetrap: Current and emerging humanized mouse and rat models for investigating prevention and cure strategies against HIV infection and associated pathologies. Retrovirology 2020, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Dzobo, K.; Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Mwapagha, L.M.; Al-Awwad, N.; Dandara, C.; Parker, M.I. Cancer Stem Cell Hypothesis for Therapeutic Innovation in Clinical Oncology? Taking the Root Out, Not Chopping the Leaf. OMICS A J. Integr. Boil. 2016, 20, 681–691. [Google Scholar] [CrossRef]
- Bozorgi, A.; Khazaei, M.; Khazaei, M.R. New Findings on Breast Cancer Stem Cells: A Review. J. Breast Cancer 2015, 18, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R. Gastric cancer stem cells: A novel therapeutic target. Cancer Lett. 2013, 338, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef]
- Dzobo, K.; Rowe, A.; Senthebane, D.A.; Almazyadi, M.A.; Patten, V.; Parker, M.I. Three-Dimensional Organoids in Cancer Research: The Search for the Holy Grail of Preclinical Cancer Modeling. OMICS J. Integr. Boil. 2018, 22, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Sánchez, E.; Santiago-López, L.; Cruz-Domínguez, V.B.; Toledo-Guzmán, M.E.; Hernández-Cueto, D.; Muñiz-Hernández, S.; Garrido, E.; De León, D.C.; García-Carrancá, A. Characterization of cervical cancer stem cell-like cells: Phenotyping, stemness, and human papilloma virus co-receptor expression. Oncotarget 2016, 7, 31943–31954. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.; Zhang, W.; Tong, C.; Kazobinka, G.; Huang, X.; Huang, Y.; Zhang, Y. Putative stem cell markers in cervical squamous cell carcinoma are correlated with poor clinical outcome. BMC Cancer 2015, 15, 785. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Vishnoi, K.; Mahata, S.; Verma, G.; Srivastava, Y.; Masaldan, S.; Roy, B.G.; Bharti, A.C.; Das, B.C. Cervical Cancer Stem Cells Selectively Overexpress HPV Oncoprotein E6 that Controls Stemness and Self-Renewal through Upregulation of HES1. Clin. Cancer Res. 2016, 22, 4170–4184. [Google Scholar] [CrossRef] [Green Version]
- Ming, X.-Y.; Fu, L.; Zhang, L.-Y.; Qin, Y.-R.; Cao, T.-T.; Chan, K.W.; Ma, S.K.Y.; Xie, D.; Guan, X.-Y. Integrin α7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat. Commun. 2016, 7, 13568. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.-S.; Li, W.-J.; Ge, D.; Zhang, P.-J.; Li, J.-J.; Lu, C.-L.; Ji, X.-D.; Guan, D.-X.; Gao, H.; Xu, L.-Y.; et al. Tumor Initiating Cells in Esophageal Squamous Cell Carcinomas Express High Levels of CD44. PLoS ONE 2011, 6, e21419. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.-X.; Mo, J.; Zhao, G.; Shu, G.; Fu, H.-L.; Zhao, W. Targeting Strategies for Renal Cell Carcinoma: From Renal Cancer Cells to Renal Cancer Stem Cells. Front. Pharmacol. 2016, 7, 423. [Google Scholar] [CrossRef] [Green Version]
- Peired, A.; Sisti, A.; Romagnani, P. Renal Cancer Stem Cells: Characterization and Targeted Therapies. Stem Cells Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.; Yang, G.; Jiang, R.; Cheng, Y.; Yang, H.; Pei, L.; Qiu, X. Cancer stem cell markers predict a poor prognosis in renal cell carcinoma: A meta-analysis. Oncotarget 2016, 7, 65862–65875. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Bellister, S.; Lü, J.; Ye, X.; Boulbès, D.R.; Tozzi, F.; Sceusi, E.; Kopetz, S.; Tian, F.; Xia, L.; et al. The requirement for freshly isolated human colorectal cancer (CRC) cells in isolating CRC stem cells. Br. J. Cancer 2014, 112, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D.; et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F.B.J. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Nat. Acad. Sci. USA 2010, 107, 3722–3727. [Google Scholar] [CrossRef] [Green Version]
- Kemper, K.; Prasetyanti, P.R.; De Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal Antibodies Against Lgr5 Identify Human Colorectal Cancer Stem Cells. Stem Cells 2012, 30, 2378–2386. [Google Scholar] [CrossRef]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ Cancer Stem Cells in Human Liver Cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Kimura, O.; Takahashi, T.; Ishii, N.; Inoue, Y.; Ueno, Y.; Kogure, T.; Fukushima, K.; Shiina, M.; Yamagiwa, Y.; Kondo, Y.; et al. Characterization of the epithelial cell adhesion molecule (EpCAM) + cell population in hepatocellular carcinoma cell lines. Cancer Sci. 2010, 101, 2145–2155. [Google Scholar] [CrossRef]
- Nie, S.; McDermott, S.P.; Deol, Y.; Tan, Z.; Wicha, M.S.; Lubman, D.M. A quantitative proteomics analysis of MCF7 breast cancer stem and progenitor cell populations. Proteomics. 2015, 15, 3772–3783. [Google Scholar] [CrossRef] [Green Version]
- A Theodoropoulos, P.; Chiotaki, R.; Polioudaki, H. Stem cell technology in breast cancer: Current status and potential applications. Stem Cells Cloning Adv. Appl. 2016, 9, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Li, C.; He, F.; Cai, Y.; Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. J. Cancer Res. Clin. Oncol. 2011, 137, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/β-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Ma, L.; Yi, D.; Yoon, J.-G.; Diercks, A.; Foltz, G.; Price, N.D.; Hood, L.E.; Tian, Q. A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc. Nat. Acad. Sci. USA 2011, 108, 1591–1596. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.Y.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Boil. 2005, 15, 494–501. [Google Scholar] [CrossRef]
- Afify, S.M.M.; Hassan, G.; Osman, A.; Calle, A.S.; Nawara, H.M.; Zahra, M.H.; El-Ghlban, S.; Mansour, H.; Alam, J.; Abu Quora, H.A.; et al. Metastasis of Cancer Stem Cells Developed in the Microenvironment of Hepatocellular Carcinoma. Bioengineering 2019, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, C.I.; Shinohara, H.; Miyamoto, K.; Yamauchi, N.; Yuda, J.; Akao, Y.; Minami, Y. Chronic myeloid leukemia stem cells and molecular target therapies for overcoming resistance and disease persistence. Int. J. Hematol. 2018, 108, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian cancer stem cell markers: Prognostic and therapeutic implications. Cancer Lett. 2012, 322, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.T.; Maitland, N.J. Prostate cancer stem cells. Eur. J. Cancer 2006, 42, 1213–1218. [Google Scholar] [CrossRef]
- Chen, X.; Li, Q.; Liu, X.; Liu, C.; Liu, R.; Rycaj, K.; Zhang, D.; Liu, B.; Jeter, C.; Calhoun-Davis, T.; et al. Defining a Population of Stem-like Human Prostate Cancer Cells That Can Generate and Propagate Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4505–4516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, X.; Rycaj, K.; Chao, H.-P.; Deng, Q.; Jeter, C.R.; Liu, C.; Honorio, S.; Li, H.; Davis, T.; et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget 2015, 6, 23959–23986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic Cancer Stem Cells. J. Clin. Oncol. 2008, 26, 2806–2812. [Google Scholar] [CrossRef]
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of Human Pancreatic Cancer Stem Cells. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2009; Volume 568, pp. 161–173. [Google Scholar]
- La Porta, C.A.M. Cancer Stem Cells: Lessons from Melanoma. Stem Cell Rev. Rep. 2008, 5, 61–65. [Google Scholar] [CrossRef]
- Kumar, D.; Gorain, M.; Kundu, G.; Kundu, G.C. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 2017, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Chinn, S.B.; Darr, O.A.; Owen, J.H.; Bellile, E.; McHugh, J.B.; Spector, M.E.; Papagerakis, S.M.; Chepeha, U.B.; Bradford, C.R.; Carey, T.E.; et al. Cancer stem cells: Mediators of tumorigenesis and metastasis in head and neck squamous cell carcinoma. Head Neck 2014, 37, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Shin, J.H.; Chen, C.-H.; Cruz, L.; Farnebo, L.; Yang, J.; Borges, P.; Kang, G.; Mochly-Rosen, D.; Sunwoo, J.B. Targeting aldehyde dehydrogenase activity in head and neck squamous cell carcinoma with a novel small molecule inhibitor. Oncotarget 2017, 8, 52345–52356. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Huang, B.; Liu, R.; Ju, X.; Zhou, Y.; Jiang, J.; Liang, W.; Shen, Y.; Li, F.; Pang, L. Expression of cancer stem cell markers and their correlation with pathogenesis in vascular tumors. Int. J. Clin. Exp. Pathol. 2015, 8, 12621–12633. [Google Scholar]
- Veselska, R.; Skoda, J.; Neradil, J. Detection of cancer stem cell markers in sarcomas. Klin. Onkol. 2012, 25, 16–20. [Google Scholar]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, N.; Mizushima, T.; Doki, Y.; Mori, M. Cancer stem cells in relation to treatment. Jpn. J. Clin. Oncol. 2018, 49, 232–237. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology (Williston Park. N. Y.) 2014, 28, 1101–1107. [Google Scholar]
- Steinbichler, T.B.; Dudas, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.-I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Boil. 2018, 53, 156–167. [Google Scholar] [CrossRef]
- Dymock, B.W. The rise of epigenetic drug discovery. Futur. Med. Chem. 2016, 8, 1523–1524. [Google Scholar] [CrossRef]
- Hsu, L.C.; Chang, W.C.; Hoffmann, I.; Duester, G. Molecular analysis of two closely related mouse aldehyde dehydrogenase genes: Identification of a role for Aldh1, but not Aldh-pb, in the biosynthesis of retinoic acid. Biochem. J. 1999, 339, 387–395. [Google Scholar] [CrossRef]
- Yoshida, A.; Rzhetsky, A.; Hsu, L.C.; Chang, C. Human aldehyde dehydrogenase gene family. JBIC J. Boil. Inorg. Chem. 1998, 251, 549–557. [Google Scholar] [CrossRef]
- Eirew, P.; Kannan, N.; Knapp, D.J.H.F.; Vailllant, F.; Emerman, J.T.; Lindeman, G.J.; Visvader, J.E.; Eaves, C.J. Aldehyde Dehydrogenase Activity Is a Biomarker of Primitive Normal Human Mammary Luminal Cells. Stem Cells 2012, 30, 344–348. [Google Scholar] [CrossRef]
- Ioannou, M.; Serafimidis, I.; Arnes, L.; Sussel, L.; Singh, S.; Vasiliou, V.; Gavalas, A. ALDH1B1 is a potential stem/progenitor marker for multiple pancreas progenitor pools. Dev. Boil. 2012, 374, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde Dehydrogenases in Cellular Responses to Oxidative/electrophilic Stress. Free Radic. Boil. Med. 2012, 56, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Vassalli, G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311. [Google Scholar] [CrossRef]
- Vogler, T.; Kriegl, L.; Horst, D.; Engel, J.; Sagebiel, S.; Schäffauer, A.J.; Kirchner, T.; Jung, A. The expression pattern of aldehyde dehydrogenase 1 (ALDH1) is an independent prognostic marker for low survival in colorectal tumors. Exp. Mol. Pathol. 2012, 92, 111–117. [Google Scholar] [CrossRef]
- Hoogen, C.V.D.; Van Der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; Pelger, R.C.; et al. High Aldehyde Dehydrogenase Activity Identifies Tumor-Initiating and Metastasis-Initiating Cells in Human Prostate Cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Ogasawara, S.; Akiba, J.; Nakayama, M.; Todoroki, K.; Ueda, K.; Sanada, S.; Suekane, S.; Noguchi, M.; Matsuoka, K.; et al. Aldehyde Dehydrogenase 1 Identifies Cells with Cancer Stem Cell-Like Properties in a Human Renal Cell Carcinoma Cell Line. PLoS ONE 2013, 8, e75463. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.K.Y.; Lee, T.K.; Zheng, B.-J.; Chan, K.W.; Guan, X.-Y.; Lee, T.K.W. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2007, 27, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Zhao, Y.; Qian, J.; Tang, J.; Zhan, X.-D. Aldehyde dehydrogenase 1 can be used as a new marker of cancer stem cells in laryngeal cancer cells in vitro. Zhonghua Zhong Liu Za Zhi (Chin. J. Oncol.) 2011, 33, 900–904. [Google Scholar]
- Afify, S.M.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, K.; Kim, S.J.; Tanei, T.; Shimazu, K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Terada, N.; Noguchi, S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009, 100, 1062–1068. [Google Scholar] [CrossRef]
- Rausch, V.; Liu, L.; Kallifatidis, G.; Baumann, B.; Mattern, J.; Gladkich, J.; Wirth, T.; Schemmer, P.; Buchler, M.W.; Zöller, M.; et al. Synergistic Activity of Sorafenib and Sulforaphane Abolishes Pancreatic Cancer Stem Cell Characteristics. Cancer Res. 2010, 70, 5004–5013. [Google Scholar] [CrossRef] [Green Version]
- Yip, N.C.; Fombon, I.S.; Liu, P.; Brown, S.; Kannappan, V.; Armesilla, A.L.; Xu, B.; Cassidy, J.; Darling, J.L.; Wang, W. Disulfiram modulated ROS–MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br. J. Cancer 2011, 104, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Boil. 2016, 23, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Lario, A.P.; García, C.B.; Elizondo, M.E.; Lobo, C. Expression of proteins associated with multidrug resistance and resistance to chemotherapy in lung cancer. Arch. Bronconeumol. (Engl. Ed.) 2007, 43, 479–484. [Google Scholar] [CrossRef]
- Leonessa, F.; Clarke, R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr. Relat. Cancer 2003, 10, 43–73. [Google Scholar] [CrossRef] [Green Version]
- Boesch, M.; Zeimet, A.G.; Rumpold, H.; Gastl, G.; Sopper, S.; Wolf, D. Drug Transporter-Mediated Protection of Cancer Stem Cells from Ionophore Antibiotics. Stem Cells Transl. Med. 2015, 4, 1028–1032. [Google Scholar] [CrossRef]
- Xiong, B.; Ma, L.; Hu, X.; Zhang, C.; Cheng, Y. Characterization of side population cells isolated from the colon cancer cell line SW480. Int. J. Oncol. 2014, 45, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.H.; Margaryan, A.; Sadée, W.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Waaga-Gasser, A.M.; Sayegh, M.H. ABCB5-Mediated Doxorubicin Transport and Chemoresistance in Human Malignant Melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.-M.; Xu, Y.; Fan, J.; Zhou, J.; Yang, X.-R.; Qiu, S.-J.; Liao, Y.; Wu, W.-Z.; Ji, Y.; Ke, A.-W.; et al. Identification of side population cells in human hepatocellular carcinoma cell lines with stepwise metastatic potentials. J. Cancer Res. Clin. Oncol. 2008, 134, 1155–1163. [Google Scholar] [CrossRef]
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lou, H.; Dean, M. Targeted therapy for cancer stem cells: The patched pathway and ABC transporters. Oncogene 2007, 26, 1357–1360. [Google Scholar] [CrossRef] [Green Version]
- Marcelletti, J.F.; Multani, P.S.; Lancet, J.E.; Baer, M.R.; Sikic, B.I. Leukemic blast and natural killer cell P-glycoprotein function and inhibition in a clinical trial of zosuquidar infusion in acute myeloid leukemia. Leuk. Res. 2009, 33, 769–774. [Google Scholar] [CrossRef]
- Tang, R.; Faussat, A.-M.; Perrot, J.-Y.; Marjanovic, Z.; Cohen, S.; Storme, T.; Morjani, H.; Legrand, O.; Marie, J.-P. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-R.; Wang, S.; Yan, S.-C.; Zhang, Y.; Nelson, P.J.; Jia, H.-L.; Qin, L.-X.; Dong, Q.-Z. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front. Oncol. 2019, 9, 9. [Google Scholar] [CrossRef]
- Strasser, A.; Harris, A.W.; Bath, M.L.; Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990, 348, 331–333. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Wang, Y.; Desgrosellier, J.S. Combined Bcl-2/Src inhibition synergize to deplete stem-like breast cancer cells. Cancer let. 2019, 457, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; De Franco, F.; Orleth, A.; Cambiaghi, V.; Giuliani, V.; Bossi, D.; Ronchini, C.; Ronzoni, S.; Muradore, I.; Monestiroli, S.; et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009, 457, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S.; Sridharan, D.; Brown, M.; Lambert, W.C.; McMahon, L.W. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mccord, A.M.; Jamal, M.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin. Cancer Res. 2009, 15, 5145–5153. [Google Scholar] [CrossRef] [Green Version]
- Gallmeier, E.; Hermann, P.C.; Mueller, M.-T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of Ataxia Telangiectasia- and Rad3 -Related Function Abrogates the In Vitro and In Vivo Tumorigenicity of Human Colon Cancer Cells Through Depletion of the CD133+ Tumor-Initiating Cell Fraction. Stem Cells 2011, 29, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Morgenstern, M.F.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; et al. Dynamic Proteomics of Individual Cancer Cells in Response to a Drug. Science 2008, 322, 1511–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, S.; Hersey, J.M.; Mellor, P.; Dai, W.; Santos-Silva, A.; Liber, D.; Luk, L.; Titley, I.; Carden, C.P.; Box, G.; et al. Ovarian cancer stem cell like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer Ther. 2011, 10, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levina, V.; Marrangoni, A.M.; Demarco, R.; Gorelik, E.; Lokshin, A.E. Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network, Tumorigenic and Metastatic Properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.-K.; Shen, N.; Chen, Y.-S.; Yang, Q.-Y.; Guo, C.-C.; Feng, B.-H.; Chen, Z.-P. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin. J. Cancer 2014, 33, 115–122. [Google Scholar] [CrossRef]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.E.; Lerner, S.P.; Chen, F.; Roh, T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2014, 517, 209–213. [Google Scholar] [CrossRef]
- Saito, Y.; Uchida, N.; Tanaka, S.; Suzuki, N.; Tomizawa-Murasawa, M.; Sone, A.; Najima, Y.; Takagi, S.; Aoki, Y.; Wake, A.; et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 2010, 28, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Vitiani, L.R.; Gulotta, G.; Dieli, F.; De Maria, R.; Stassi, G. Bone Morphogenetic Protein 4 Induces Differentiation of Colorectal Cancer Stem Cells and Increases Their Response to Chemotherapy in Mice. Gastroenterology 2011, 140, 297–309.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Chang, C.-C.; Chiang, C.-C.; Chen, W.-M.; Hung, S.-C. Silibinin suppresses the maintenance of colorectal cancer stem-like cells by inhibiting PP2A/AKT/mTOR pathways. J. Cell. Biochem. 2011, 113, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 decades of development. Blood 2009, 113, 3655–3665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhang, J.; Wang, Z.-Y.; Chen, S.-J.; Chen, Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: A paradigm of synergistic molecular targeting therapy. Philos. Trans. R Soc. B Boil. Sci. 2007, 362, 959–971. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Pour, L.; Hájek, R.; Buchler, T.; Maisnar, V.; Smolej, L. Angiogenesis and antiangiogenic cancer therapy. Vnitrni Lek. 2004, 50, 930–938. [Google Scholar]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Pandya, N.; Dhalla, N.S.; Santani, D.D. Angiogenesis—A new target for future therapy. Vasc. Pharmacol. 2006, 44, 265–274. [Google Scholar] [CrossRef]
- Furuya, M. Complexity of tumor vasculature and molecular targeting therapies. Front. Biosci. 2011, 3, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis—characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciuti, B.; Foglietta, J.; Bianconi, V.; Sahebkar, A.; Pirro, M. Enzymes involved in tumor-driven angiogenesis: A valuable target for anticancer therapy. Semin. Cancer Boil. 2019, 56, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Kim, S.; Hong, B.-J.; Lee, C.-J.; Kim, Y.-E.; Bok, S.; Oh, J.-M.; Gwak, S.-H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Tanriover, G.; Aytac, G. Mutualistic Effects of the Myeloid-Derived Suppressor Cells and Cancer Stem Cells in the Tumor Microenvironment. Crit. Rev. Oncog. 2019, 24, 61–67. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Wang, Y.; Han, W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: A potential and curable approach for cancer treatment. Protein Cell 2017, 9, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Espinoza, I.; Pochampally, R.R.; Watabe, K.; Xing, F. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. OncoTargets Ther. 2013, 6, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Liu, Z.; Oh, D.-Y.; Thiele, C.J. MYCN and the epigenome. Front. Oncol. 2013, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Jiang, J.-H.; Zhang, J.; Yang, H.-J.; Yang, F.-Q.; Qi, Y.-P.; Zhong, Y.-P.; Su, J.; Yang, R.-R.; Li, L.-Q.; et al. COX-2 Promotes Migration and Invasion by the Side Population of Cancer Stem Cell-Like Hepatocellular Carcinoma Cells. Medicine 2015, 94, e1806. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Şenbabaoğlu, Y.; Conley, S.J.; et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol. Cancer Ther. 2015, 14, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, T.S.; Riz, I.; Yang, W.; Wakabayashi, Y.; DePalma, L.; Chang, Y.-T.; Peng, W.; Zhu, J.; Hawley, R. Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am. J. Hematol. 2013, 88, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J. Cell. Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The Notch Ligands Dll4 and Jagged1 Have Opposing Effects on Angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Owen, J.L.; Zadeh, M. Macrophages and chemokines as mediators of angiogenesis. Front. Physiol. 2013, 4, 159. [Google Scholar] [CrossRef] [Green Version]
- Meier, K.; Lehr, C.-M.; Daum, N. Differentiation potential of human pancreatic stem cells for epithelial- and endothelial-like cell types. Ann. Anat. Anat. Anz. 2009, 191, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.W.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Grillet, F.; Bayet, E.; Villeronce, O.; Zappia, L.; Lagerqvist, E.L.; Lunke, S.; Charafe-Jauffret, E.; Pham, K.; Molck, C.; Rolland, N.; et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 2017, 66, 1802–1810. [Google Scholar] [CrossRef] [Green Version]
- Burgess, D.J. Breast cancer: Circulating and dynamic EMT. Nat. Rev. Cancer 2013, 13, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.W.; Bruns, C.J. Cancer stem cells and angiogenesis. Int. J. Dev. Boil. 2011, 55, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S. Stem Cell-like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, K.; Grandela, C.; Medema, J.P. Molecular identification and targeting of colorectal cancer stem cells. Oncotarget 2010, 1, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yuan, B.; Zhang, H.; Li, H. Human epithelial ovarian cancer cells expressing CD105, CD44 and CD106 surface markers exhibit increased invasive capacity and drug resistance. Oncol. Lett. 2019, 17, 5351–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chen, L.; Deng, H.; Xu, B.; Li, M.; Zheng, X.; Wu, C.; Jiang, J.-T. Epithelial-to-mesenchymal transition in human esophageal cancer associates with tumor progression and patient’s survival. Int. J. Clin. Exp. Pathol. 2014, 7, 6943–6949. [Google Scholar] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore) 2016, 95, S20–S25. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sainz, B. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Boil. 2018, 53, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Ni, W.; Cui, C.; Qi, W.; Piao, L.; Xuan, Y. Identification of LETM1 as a marker of cancer stem-like cells and predictor of poor prognosis in esophageal squamous cell carcinoma. Hum. Pathol. 2018, 81, 148–156. [Google Scholar] [CrossRef]
- Gupta, P.B.; Önder, T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Bure, I.V.; Nemtsova, M.V.; Zaletaev, D.V. Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Zhang, H.-S.; Liu, Y.; Zhang, Z.-G.; Du, G.-Y.; Li, H.; Yu, X.-Y.; Huang, Y. Loss of TET1 facilitates DLD1 colon cancer cell migration via H3K27me3-mediated down-regulation of E-cadherin. J. Cell. Physiol. 2017, 233, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Hill, C.S. Beyond TGFβ: Roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [Green Version]
- Ganai, S.A. Histone deacetylase inhibitors modulating non-epigenetic players: The novel molecular targets for therapeutic intervention. Curr. Drug Targets 2018, 17, 1. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, J.; Han, S.; Kim, J.H.; Mani, R.-S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Kleer, C.G.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Boil. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Guan, T.; Dominguez, C.X.; Amezquita, R.A.; Laidlaw, B.J.; Cheng, J.; Henao-Mejia, J.; Williams, A.; Flavell, R.A.; Lu, J.; Kaech, S.M. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8+ T cell fates. J. Exp. Med. 2018, 215, 1153–1168. [Google Scholar] [CrossRef]
- Tellez, C.S.; Juri, D.E.; Do, K.; Bernauer, A.M.; Thomas, C.L.; Damiani, L.A.; Tessema, M.; Leng, S.; Belinsky, S.A. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011, 71, 3087–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menéndez, J.; Alarcón, T. Metabostemness: A New Cancer Hallmark. Front. Oncol. 2014, 4, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouyssegur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Nakada, D. Venetolax with Azacitidine Drains Fuel from AML Stem Cells. Cell Stem Cell 2019, 24, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Amsalem, Z.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers 2020, 12, 1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoda, J.; Borankova, K.; Jansson, P.; Huang, M.L.-H.; Veselska, R.; Richardson, D.R. Pharmacological targeting of mitochondria in cancer stem cells: An ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019, 139, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.-S.; Jeong, Y.J.; Jeong, S.H.; Heo, H.J.; Kim, H.K.; Bae, K.B.; Park, Y.-H.; Kim, S.-U.; Kim, J.-M.; Kim, N.; et al. FOXM1-Induced PRX3 Regulates Stemness and Survival of Colon Cancer Cells via Maintenance of Mitochondrial Function. Gastroenterology 2015, 149, 1006–1016.e9. [Google Scholar] [CrossRef] [PubMed]
- Isayev, O.; Rausch, V.; Bauer, N.; Liu, L.; Fan, P.; Zhang, Y.; Gladkich, J.; Nwaeburu, C.C.; Mattern, J.; Mollenhauer, M.; et al. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget 2014, 5, 5177–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Boil. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Borley, J.; Brown, R. Epigenetic mechanisms and therapeutic targets of chemotherapy resistance in epithelial ovarian cancer. Ann. Med. 2015, 47, 359–369. [Google Scholar] [CrossRef]
- Arrowsmith, C.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [Green Version]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.-J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Boil. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolny, E.; Braszewska-Zalewska, A.; Kroczek, D.; Hasterok, R. Histone H3 and H4 acetylation patterns are more dynamic than those of DNA methylation in Brachypodium distachyon embryos during seed maturation and germination. Protoplasma 2017, 254, 2045–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. eLife 2018, 7, 34081. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Zhou, K.; Wu, T.; Zhao, B.S.; Sun, M.; Chen, Z.; Deng, X.; Xiao, G.; Auer, F.; et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 2019, 567, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, R.; Cui, X.; Zha, L.; Yu, L.; Shi, H.; Xue, B. Histone Deacetylase 1 (HDAC1) Negatively Regulates Thermogenic Program in Brown Adipocytes via Coordinated Regulation of Histone H3 Lysine 27 (H3K27) Deacetylation and Methylation. J. Boil. Chem. 2016, 291, 4523–4536. [Google Scholar] [CrossRef] [Green Version]
- Möller, M.; Schotanus, K.; Soyer, J.L.; Haueisen, J.; Happ, K.; Stralucke, M.; Happel, P.; Smith, K.M.; Connolly, L.R.; Freitag, M.; et al. Destabilization of chromosome structure by histone H3 lysine 27 methylation. PLoS Genet. 2019, 15, e1008093. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Wolberger, C. How does the histone code work? Biochem. Cell. Biol. 2005, 83, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Cloney, R. Gene regulation: Optical control of epigenetics. Nat. Rev. Genet. 2016, 17, 254. [Google Scholar] [PubMed]
- Harvey, Z.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2017, 69, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Lee, J.E.; Kim, L.K.; Kim, T. Epigenetic memory in gene regulation and immune response. BMB Rep. 2019, 52, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M. CpG Islands. In Encyclopedia of Genetics; Elsevier BV: Amsterdam, The Netherlands, 2001; p. 477. [Google Scholar]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani-Fujita, N.; Fujita, T.; Aoike, A.; Osifchin, N.E.; Robbins, P.D.; Sakai, T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene 1993, 8, 8. [Google Scholar]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Boil. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef]
- Corso-Díaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res. 2018, 65, 1–27. [Google Scholar] [CrossRef]
- Collinson, A.; Collier, A.; Morgan, N.P.; Sienerth, A.; Chandra, T.; Andrews, S.; Rugg-Gunn, P.J. Deletion of the Polycomb-Group Protein EZH2 Leads to Compromised Self-Renewal and Differentiation Defects in Human Embryonic Stem Cells. Cell Rep. 2016, 17, 2700–2714. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef]
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2014, 263, 22–35. [Google Scholar] [CrossRef]
- Yi, J.M.; Tsai, H.-C.; Glöckner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103. [Google Scholar] [CrossRef] [Green Version]
- Tabu, K.; Sasai, K.; Kimura, T.; Wang, L.; Aoyanagi, E.; Kohsaka, S.; Tanino, M.; Nishihara, H.; Tanaka, K. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008, 18, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Gorodetska, I.V.; Lukiyanchuk, V.; Peitzsch, C.; Kozeretska, I.; Dubrovska, A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int. J. Cancer 2019, 145, 2974–2985. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Zhu, X. The Wnt/beta-catenin pathway regulates self-renewal of cancer stem-like cells in human gastric cancer. Mol. Med. Rep. 2012, 5, 1191–1196. [Google Scholar]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Zhu, C.; Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F. Therapeutic approaches targeting cancer stem cells. J. Cancer Res. Ther. 2018, 14, 1469. [Google Scholar] [CrossRef]
- Lai, E. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Zhang, J.; Shao, X.; Sun, H.; Liu, K.; Ding, Z.; Chen, J.; Fang, L.; Su, W.; Hong, Y.; Li, H.; et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 2016, 7, 61036–61053. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2009, 29, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; i Altaba, A.R. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Steinway, S.N.; Zañudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P.; Albert, R. Network Modeling of TGF Signaling in Hepatocellular Carcinoma Epithelial-to-Mesenchymal Transition Reveals Joint Sonic Hedgehog and Wnt Pathway Activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.-X.; Wang, S.; Zhao, H.; Liu, N.; Chen, D.; Sun, M.; Zheng, J. Sonic hedgehog signaling may promote invasion and metastasis of oral squamous cell carcinoma by activating MMP-9 and E-cadherin expression. Med. Oncol. 2014, 31, 1–8. [Google Scholar] [CrossRef]
- Suzuki, H.; Watkins, D.N.; Jair, K.-W.; Schuebel, K.E.; Markowitz, S.D.; Chen, W.D.; Pretlow, T.P.; Yang, B.; Akiyama, Y.; Van Engeland, M.; et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 2004, 36, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Rao, M.; Humphries, A.E.; Hong, J.A.; Liu, F.; Yang, M.; Caragacianu, D.; Schrump, D.S. Tobacco Smoke Induces Polycomb-Mediated Repression of Dickkopf-1 in Lung Cancer Cells. Cancer Res. 2009, 69, 3570–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, P.; Nganga, A.J.; Moran-Giuati, J.; Szafranek, A.; Johnson, T.R.; Bigelow, A.J.; Houde, C.M.; Avet-Loiseau, H.; Smiraglia, D.J.; Ersing, N.; et al. Loss of the SMRT/NCoR2 Corepressor Correlates with JAG2 Overexpression in Multiple Myeloma. Cancer Res. 2009, 69, 4380–4387. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Gong, L.; Xu, J.; Mou, Y.; Xu, X.; Qian, Z. The clinicopathological significance of HES1 promoter hypomethylation in patients with colorectal cancer. OncoTargets Ther. 2017, 10, 5827–5834. [Google Scholar] [CrossRef] [Green Version]
- Benoit, Y.D.; Laursen, K.B.; Witherspoon, M.S.; Lipkin, S.M.; Gudas, L.J. Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J. Cell. Physiol. 2013, 228, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Ranganathan, P.; Tzimas, C.; Weaver, K.L.; Jin, K.; Astudillo, L.; Zhou, W.; Zhu, X.; Li, B.; Robbins, D.J.; et al. Notch Represses Transcription by PRC2 Recruitment to the Ternary Complex. Mol. Cancer Res. 2017, 15, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- McKenna, E.S.; Tamayo, P.; Cho, Y.-J.; Tillman, E.J.; Mora-Blanco, E.L.; Sansam, C.G.; Koellhoffer, E.C.; Pomeroy, S.L.; Roberts, C.W.M. Epigenetic inactivation of the tumor suppressor BIN1 drives proliferation of SNF5-deficient tumors. Cell Cycle 2012, 11, 1956–1965. [Google Scholar] [CrossRef] [Green Version]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell. Biol. 2010, 12, 132–142. [Google Scholar]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalak, E.M.; Visvader, J.E. Dysregulation of histone methyltransferases in breast cancer – Opportunities for new targeted therapies? Molecular Oncol. 2016, 10, 1497–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Cozzio, A.; Passegué, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef] [Green Version]
- Ono, R.; Masuya, M.; Nakajima, H.; Enomoto, Y.; Miyata, E.; Nakamura, A.; Ishii, S.; Suzuki, K.; Shibata-Minoshima, F.; Katayama, N.; et al. Plzf drives MLL-fusion–mediated leukemogenesis specifically in long-term hematopoietic stem cells. Blood 2013, 122, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical Maintenance of MLL Myeloid Leukemia Stem Cells Employs a Transcriptional Program Shared with Embryonic Rather Than Adult Stem Cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Benetatos, L.; Vartholomatos, G. On the potential role of DNMT1 in acute myeloid leukemia and myelodysplastic syndromes: Not another mutated epigenetic driver. Ann. Hematol. 2016, 95, 1571–1582. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; A Goodell, M. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2016, 7, a030320. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, B.-R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, M.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2017, 71, 110–116. [Google Scholar] [CrossRef]
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon Cancer Stem Cells: Promise of Targeted Therapy. Gastroenterology 2010, 138, 2151–2162. [Google Scholar] [CrossRef]
- Hammoud, S.S.; Cairns, B.R.; Jones, D.A. Epigenetic regulation of colon cancer and intestinal stem cells. Curr. Opin. Cell Boil. 2013, 25, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.M.; Szyf, M. Human DNA methyltransferase gene DNMT1 is regulated by the APC pathway. Carcinog. 2003, 24, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.-J.; Wang, H.-X.; Zheng, Y.-Y.; Cao, Y.-Q.; Wu, X.; Zhou, X.; Dong, S.-X. APC hypermethylation for early diagnosis of colorectal cancer: A meta-analysis and literature review. Oncotarget 2017, 8, 46468–46479. [Google Scholar] [CrossRef] [Green Version]
- Paschall, A.V.; Liu, K. Epigenetic and Immune Regulation of Colorectal Cancer Stem Cells. Curr. Color. Cancer Rep. 2015, 11, 414–421. [Google Scholar] [CrossRef]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Xia, H.; Cao, J.; Li, Q.; Lv, Y.; Jia, W.; Ren, W.; Cheng, Q.; Song, X.; Xu, G. Hepatocellular Carcinoma-propagating Cells are Detectable by Side Population Analysis and Possess an Expression Profile Reflective of a Primitive Origin. Sci. Rep. 2016, 6, 34856. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Boesch, M.; Zeimet, A.G.; Reimer, D.; Schmidt, S.; Gastl, G.; Parson, W.; Spoeck, F.; Hatina, J.; Wolf, D.; Sopper, S. The side population of ovarian cancer cells defines a heterogeneous compartment exhibiting stem cell characteristics. Oncotarget 2014, 5, 7027–7039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Hatina, J.; Boesch, M.; Sopper, S.; Kripnerova, M.; Wolf, D.; Reimer, D.; Marth, C.; Zeimet, A.G. Ovarian Cancer Stem Cell Heterogeneity. Single Mol. Single Cell Seq. 2019, 1139, 201–221. [Google Scholar] [CrossRef]
- Smith, L.M.; Nesterova, A.; Ryan, M.C.; Duniho, S.; Jonas, M.; Anderson, M.; Zabinski, R.F.; Sutherland, M.K.; Gerber, H.-P.; Van Orden, K. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br. J. Cancer 2008, 99, 100–109. [Google Scholar] [CrossRef]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Krampitz, G.W.; George, B.M.; Willingham, S.B.; Volkmer, J.-P.; Weiskopf, K.; Jahchan, N.; Newman, A.M.; Sahoo, D.; Zemek, A.J.; Yanovsky, R.L.; et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc. Nat. Acad. Sci. USA 2016, 113, 4464–4469. [Google Scholar] [CrossRef] [Green Version]
- Piccione, E.C.; Juarez, S.; Tseng, S.; Liu, J.; Stafford, M.; Narayanan, C.; Wang, L.; Weiskopf, K.; Majeti, R. SIRPα-Antibody Fusion Proteins Selectively Bind and Eliminate Dual Antigen-Expressing Tumor Cells. Clin. Cancer Res. 2016, 22, 5109–5119. [Google Scholar] [CrossRef] [Green Version]
- Boesch, M.; Onder, L.; Cheng, H.-W.; Novkovic, M.; Mörbe, U.; Sopper, S.; Gastl, G.; Jochum, W.; Ruhstaller, T.; Knauer, M.; et al. Interleukin 7-expressing fibroblasts promote breast cancer growth through sustenance of tumor cell stemness. OncoImmunology 2018, 7, e1414129. [Google Scholar] [CrossRef] [Green Version]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Sallman, D.A.; Donnellan, W.B.; Asch, A.S.; Lee, D.J.; Al Malki, M.; Marcucci, G.; Pollyea, D.A.; Kambhampati, S.; Komrokji, R.S.; Van Elk, J.; et al. The first-in-class anti-CD47 antibody Hu5F9-G4 is active and well tolerated alone or with azacitidine in AML and MDS patients: Initial phase 1b results. J. Clin. Oncol. 2019, 37, 7009. [Google Scholar] [CrossRef]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer Stem Cells: The Promise and the Potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, S.S.; Yu, Y.; Nautiyal, J.; Patel, B.B.; Majumdar, A.P. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol. Cancer 2010, 9, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, H.-J.; Kahn, M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014, 105, 1087–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; Kouji, H.; et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2013, 31, 2501. [Google Scholar] [CrossRef]
- Jang, G.-B.; Hong, I.-S.; Kim, R.-J.; Lee, S.-Y.; Park, S.-J.; Lee, E.-S.; Park, J.H.; Yun, C.-H.; Chung, J.-U.; Lee, K.-J. Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, H.; Park, K.; Choi, Y.-K.; Nam, J.; Hong, I. CWP232228 targets liver cancer stem cells through Wnt/β-catenin signaling: A novel therapeutic approach for liver cancer treatment. Oncotarget 2016, 7, 20395–20409. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.J.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.-L.; Zhang, L.; Huang, C.-F.; Ma, S.-R.; Bu, L.-L.; Liu, J.-F.; Yu, G.-T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhang, N.; Wang, X.; Li, Y.; Qi, W.; Zhang, H.; Li, Z.; Yang, Q. Hedgehog pathway is involved in nitidine chloride induced inhibition of epithelial-mesenchymal transition and cancer stem cells-like properties in breast cancer cells. Cell Biosci. 2016, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.-I.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Nat. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Rayes, B.F.; Shahda, S.; Starodub, A.; O’Neil, B.H.; Hanna, W.T.; Shaib, W.L.; Oh, C.; Li, W.; Li, Y.; Borodyansky, L.; et al. A phase Ib extension study of cancer stemness inhibitor BB608 (napabucasin) in combination with gemcitabine and nab-paclitaxel (nab-PTX) in patients (pts) with metastatic pancreatic cancer. J. Clin. Oncol. 2016, 34, 4128. [Google Scholar] [CrossRef]
- Langleben, A.; Supko, J.G.; Hotte, S.J.; Batist, G.; Hirte, H.W.; Rogoff, H.; Li, Y.; Li, W.; Kerstein, D.; Leggett, D.; et al. A dose-escalation phase I study of a first-in-class cancer stemness inhibitor in patients with advanced malignancies. J. Clin. Oncol. 2013, 31, 2542. [Google Scholar] [CrossRef]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus placebo in refractory advanced colorectal cancer: A randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Hubbard, J.M.; Grothey, A. Napabucasin: An Update on the First-in-Class Cancer Stemness Inhibitor. Drugs 2017, 77, 1091–1103. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Graupera, M.; Vanhaesebroeck, B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016, 6, 1090–1105. [Google Scholar] [CrossRef] [Green Version]
- Kolev, V.N.; Wright, Q.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR Dual Inhibitor VS-5584 Preferentially Targets Cancer Stem Cells. Cancer Res. 2014, 75, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naujokat, C.; Steinhart, R. Salinomycin as a Drug for Targeting Human Cancer Stem Cells. J. Biomed. Biotechnol. 2012, 2012, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Xia, G.; Dong, S.; Jiang, Z.-X.; Chen, M. An iTEP-salinomycin nanoparticle that specifically and effectively inhibits metastases of 4T1 orthotopic breast tumors. Biomaterials 2016, 93, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N.; Hillan, K.J.; Gerber, H.-P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Penson, R.T.; Huang, H.Q.; Wenzel, L.B.; Monk, B.J.; Stockman, S.; Long, H.J.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; et al. Bevacizumab for advanced cervical cancer: Patient-reported outcomes of a randomised, phase 3 trial (NRG Oncology-Gynecologic Oncology Group protocol 240). Lancet Oncol. 2015, 16, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.-G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Aalders, K.C.; Tryfonidis, K.; Senkus-Konefka, E.; Cardoso, F. Anti-angiogenic treatment in breast cancer: Facts, successes, failures and future perspectives. Cancer Treat. Rev. 2017, 53, 98–110. [Google Scholar] [CrossRef]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Négrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, S.-M.; Chang, H.-J.; Kim, B.-W.; Lee, Y.S.; Park, C.-S.; Park, K.-C.; Chang, H.-S. SoLAT (Sorafenib Lenvatinib alternating treatment): A new treatment protocol with alternating Sorafenib and Lenvatinib for refractory thyroid Cancer. BMC Cancer 2018, 18, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Cao, F.; Xing, W.; Si, T.; Yu, H.; Yang, X.; Guo, Z. Efficacy of cryoablation combined with sorafenib for the treatment of advanced renal cell carcinoma. Int. J. Hyperth. 2019, 36, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgebaly, A.S.; Menshawy, A.; El Ashal, G.; Osama, O.; Ghanem, E.; Omar, A.; Negida, A. Sunitinib alone or in combination with chemotherapy for the treatment of advanced breast cancer: A systematic review and meta-analysis. Breast Dis. 2016, 36, 91–101. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Centanni, M.; Virili, C.; Miccoli, M.; Ferrari, P.; Ruffilli, I.; Ragusa, F.; Antonelli, A.; Fallahi, P.; Martinaa, C.M.F.S. Sunitinib in the Treatment of Thyroid Cancer. Curr. Med. Chem. 2019, 26, 963–972. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Pons, F.; Bellmunt, J. Sunitinib malate in the treatment of urothelial cancer. Expert Opin. Investig. Drugs 2013, 23, 115–124. [Google Scholar] [CrossRef]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramnath, N.; Creaven, P.J. Matrix metalloproteinase inhibitors. Curr. Oncol. Rep. 2004, 6, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Li, M.; Luo, T.; Yin, Y.; Jiang, Y. Matrix metalloproteinases in tumorigenesis: An evolving paradigm. Cell. Mol. Life Sci. 2011, 68, 3853–3868. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix Metalloproteinase Inhibitors and Cancer--Trials and Tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Tlatli, R.; El Ayeb, M. MMP inhibitors and cancer treatment trials, limitations and hopes for the future. Arch. l’Institut Pasteur Tunis 2013, 90, 3–21. [Google Scholar]
- Díaz, N.; Suárez, D. Molecular Dynamics Studies of Matrix Metalloproteases. Breast Cancer 2017, 1579, 111–134. [Google Scholar]
- Ray, J.M.; Stetler-Stevenson, W.G. The role of matrix metalloproteases and their inhibitors in tumour invasion, metastasis and angiogenesis. Eur Respir J. 1994, 7, 2062–2072. [Google Scholar]
- Vartak, D.G.; Gemeinhart, R.A. Matrix metalloproteases: Underutilized targets for drug delivery. J. Drug Target. 2007, 15, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors — Novel strategies bring new prospects. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Meisel, J.E.; Chang, M. Selective small-molecule inhibitors as chemical tools to define the roles of matrix metalloproteinases in disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Tang, J.; Guo, F. Identification of Inhibitors of MMPS Enzymes via a Novel Computational Approach. Int. J. Boil. Sci. 2018, 14, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lu, Y.-T.; Sun, Y.; Shi, Z.-H.; Li, N.-G.; Tang, Y.-P.; Duan, J.-A. Recent opportunities in matrix metalloproteinase inhibitor drug design for cancer. Expert Opin. Drug Discov. 2017, 13, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neuro Oncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis-Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [Green Version]
- Gaggianesi, M.; Turdo, A.; Chinnici, A.; Lipari, E.; Apuzzo, T.; Benfante, A.; Sperduti, I.; Di Franco, S.; Meraviglia, S.; Presti, E.L.; et al. IL4 Primes the Dynamics of Breast Cancer Progression via DUSP4 Inhibition. Cancer Res. 2017, 77, 3268–3279. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Perez, R.P.; Yardley, D.; Han, L.K.; Reuben, J.M.; Gao, H.; McCanna, S.; Butler, B.; Ruffini, P.A.; Liu, Y. A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C.; Lefoulon, F. Anti-Cancer Stem-like Cell Compounds in Clinical Development—An Overview and Critical Appraisal. Front. Oncol. 2016, 6, 115. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Farnie, G.; Bundred, N.J.; Simoes, B.M.; Shergill, A.; Landberg, G.; Howell, S.J.; Clarke, R.B. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin. Cancer Res. 2013, 19, 643–656. [Google Scholar] [CrossRef] [Green Version]
- Gassenmaier, M.; Chen, D.; Buchner, A.; Henkel, L.; Schiemann, M.; Mack, B.; Schendel, D.J.; Zimmermann, W.; Pohla, H. CXC Chemokine Receptor 4 is Essential for Maintenance of Renal cell Carcinoma-Initiating Cells and Predicts Metastasis. Stem Cells 2013, 31, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, F.; Cojoc, M.; Kurth, I.; Melin, N.; Bouchez, L.C.; Dubrovska, A.; Peitzsch, C. CXCR4 as biomarker for radioresistant cancer stem cells. Int. J. Radiat. Boil. 2014, 90, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 2: Efficacy, Safety, and Other Clinical Considerations. Pharm. Ther. 2017, 42, 452. [Google Scholar]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 4508794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abril-Rodriguez, G.; Ribas, A. SnapShot: Immune Checkpoint Inhibitors. Cancer Cell 2017, 31, 848–848.e1. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef]
- Mittica, G.; Genta, S.; Aglietta, M.; Valabrega, G. Immune Checkpoint Inhibitors: A New Opportunity in the Treatment of Ovarian Cancer? Int. J. Mol. Sci. 2016, 17, 1169. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.J.E.; Carlos, G.; Wakade, D.; Byth, K.; Kong, B.Y.; Chou, S.; Carlino, M.S.; Kefford, R.; Peñas, P.F. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: A single-institution cohort. J. Am. Acad. Dermatol. 2016, 74, 455–461.e1. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.-L.; Li, Z.-Q.; Zhang, G. Research Advances on Programmed Cell Death Receptor-1 Antibody in the Treatment of Lymphoma—Review. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2019, 27, 2019–2023. [Google Scholar]
- Zimmerman, M.P.; Mehr, S.R. Targeted programmed cell death in lung cancer treatment. Am. J. Manag. Care 2014, 20, E3. [Google Scholar] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Liu, M.; Su, J.; Zhang, P.; Tang, F.; Ye, P.; Devenport, M.; Wang, X.; Zhang, Y.; Liu, Y.; et al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. 2018, 28, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zheng, P. How Does an Anti-CTLA-4 Antibody Promote Cancer Immunity? Trends Immunol. 2018, 39, 953–956. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; Mandalà, M.; Del Vecchio, M.; Gogas, H.; Arance, A.; Cowey, C.L.; Dalle, S.; Schenker, M.; Sileni, V.C.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Gao, X.; McDermott, D.F. Ipilimumab in combination with nivolumab for the treatment of renal cell carcinoma. Expert Opin. Boil. Ther. 2018, 18, 947–957. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.N.A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, S.H.; Heo, Y.-S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Medeiros, L.J.; Young, K.H. Cancer Immunotherapy in Diffuse Large B-Cell Lymphoma. Front. Oncol. 2018, 8, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amraee, A.; Evazi, M.R.; Shakeri, M.; Roozbeh, N.; Ghazanfarpour, M.; Ghorbani, M.; Ansari, J.; Darvish, L. Efficacy of nivolumab as checkpoint inhibitor drug on survival rate of patients with relapsed/refractory classical Hodgkin lymphoma: A meta-analysis of prospective clinical study. Clin. Transl. Oncol. 2019, 21, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Bair, S.M.; Strelec, L.E.; Feldman, T.A.; Ahmed, G.; Armand, P.; Shah, N.N.; Singavi, A.N.; Reddy, N.; Khan, N.; Andreadis, C.; et al. Outcomes and Toxicities of Programmed Death-1 (PD-1) Inhibitors in Hodgkin Lymphoma Patients in the United States: A Real-World, Multicenter Retrospective Analysis. Oncologist 2019, 24, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchi, L.; Sawas, A.; Deng, C.; Amengual, J.E.; Colbourn, D.S.; Lichtenstein, E.A.; Khan, K.A.; Schwartz, L.H.; Connor, O.J.O. High rate of complete responses to immune checkpoint inhibitors in patients with relapsed or refractory Hodgkin lymphoma previously exposed to epigenetic therapy. J. Hematol. Oncol. 2016, 9, 132. [Google Scholar] [CrossRef] [Green Version]
- Khunger, M.; Rakshit, S.; Pasupuleti, V.; Hernandez, A.V.; Mazzone, P.; Stevenson, J.; Pennell, N.A.; Velcheti, V. Incidence of Pneumonitis with Use of Programmed Death 1 and Programmed Death-Ligand 1 Inhibitors in Non-Small Cell Lung Cancer. Chest 2017, 152, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; Salem, J.-E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune Checkpoint Inhibition Overcomes ADCP-Induced Immunosuppression by Macrophages. Cell 2018, 175, 442–457.e23. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, P.; Wang, C.; Xu, N.; Xu, A.; Xu, Y.; Sadahira, T.; Araki, M.; Wada, K.; Matsuura, E.; et al. Synergistic effects of the immune checkpoint inhibitor CTLA-4 combined with the growth inhibitor lycorine in a mouse model of renal cell carcinoma. Oncotarget 2017, 8, 21177–21186. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, H.; Fujieda, K.; Miyashita, A.; Fukushima, S.; Ikeda, T.; Kubo, Y.; Senju, S.; Ihn, H.; Nishimura, Y.; Oshiumi, H. Combined Blockade of IL6 and PD-1/PD-L1 Signaling Abrogates Mutual Regulation of Their Immunosuppressive Effects in the Tumor Microenvironment. Cancer Res. 2018, 78, 5011–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Lee, Y.; Shin, J.H.; Longmire, M.; Wang, H.; Kohrt, H.E.; Chang, H.Y.; Sunwoo, J.B. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell-Mediated Immunity by Selective Constitutive and Inducible Expression of PD-L1. Clin. Cancer Res. 2016, 22, 3571–3581. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 Expression in Breast and Colon Cancer Stem Cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef]
- Bruttel, V.S.; Wischhusen, J.; Wischhusen, J. Cancer Stem Cell Immunology: Key to Understanding Tumorigenesis and Tumor Immune Escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, C.; Ciummo, S.L.; Cipollone, G.; Caputo, S.; Bellone, M.; Di Carlo, E. Interleukin-30/IL27p28 Shapes Prostate Cancer Stem-like Cell Behavior and Is Critical for Tumor Onset and Metastasization. Cancer Res 2018, 78, 2654–2668. [Google Scholar] [CrossRef] [Green Version]
- >Szaryńska, M.; Olejniczak, A.; Kobiela, J.; Łaski, D.; Śledziński, Z.; Kmieć, Z. Cancer stem cells as targets for DC-based immunotherapy of colorectal cancer. Sci. Rep. 2018, 8, 12042. [Google Scholar] [CrossRef]
- Aliru, M.L.; Schoenhals, E.J.; Venkatesulu, B.P.; Anderson, C.C.; Barsoumian, H.B.; Younes, A.I.; Mahadevan, L.S.K.; Soeung, M.; Aziz, K.E.; Welsh, J.W.; et al. Radiation therapy and immunotherapy: What is the optimal timing or sequencing? Immunotherapy 2018, 10, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Platten, M. How to integrate immunotherapy into standard of care in glioblastoma. Neuro Oncol. 2019, 21, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.-C.; Guo, Y.-L.; Liu, Y.; Dai, H.-R.; Wang, Y.; Lv, H.-Y.; Huang, J.-H.; Yang, Q.-M.; Han, W. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-L.; Feng, K.-C.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.-M.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor–Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2017, 24, 1277–1286. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Guo, M.; Wang, Y.; Zhang, Y.; Yang, J.; Guo, Y.; Dai, H.; Yu, C.; Sun, Q.; Qiao, J.; et al. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.H.; Kim, B.W.; Song, K.-H.; Cho, H.; Lee, Y.-H.; Kim, J.H.; Chung, J.-Y.; Kim, J.-H.; Hewitt, S.M.; Seong, S.-Y.; et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J. Clin. Investig. 2012, 122, 4077–4093. [Google Scholar] [CrossRef] [Green Version]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [Green Version]
- Negmeldin, A.T.; Knoff, J.R.; Pflum, M.K.H. The structural requirements of histone deacetylase inhibitors: C4-modified SAHA analogs display dual HDAC6/HDAC8 selectivity. Eur. J. Med. Chem. 2018, 143, 1790–1806. [Google Scholar] [CrossRef] [PubMed]
- Nieto, Y.; Valdez, B.C.; Thall, P.F.; Jones, R.B.; Wei, W.; Myers, A.; Hosing, C.; Ahmed, S.; Popat, U.; Shpall, E.J.; et al. Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer 2016, 122, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Luchenko, V.; Peer, C.J.; Ghafourian, K.; Reynolds, J.; Draper, D.; Frye, R.; Woo, S.; Venzon, D.; Wright, J.; et al. Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin. Cancer Res. 2013, 19, 4499–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouladi, M.; Furman, W.L.; Chin, T.; Freeman, B.B.; Dudkin, L.; Stewart, C.F.; Krailo, M.D.; Speights, R.; Ingle, A.M.; Houghton, P.J.; et al. Phase I Study of Depsipeptide in Pediatric Patients With Refractory Solid Tumors: A Children’s Oncology Group Report. J. Clin. Oncol. 2006, 24, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.E.; Eom, J.I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK–ULK1-Mediated Autophagy Confers Resistance to BET Inhibitor JQ1 in Acute Myeloid Leukemia Stem Cells. Clin. Cancer Res. 2016, 23, 2781–2794. [Google Scholar] [CrossRef] [Green Version]
- Wen, N.; Guo, B.; Zheng, H.; Xu, L.; Liang, H.; Wang, Q.; Wang, D.; Chen, X.; Zhang, S.; Li, Y.; et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 55, 879–895. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Futur. Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.-L.; Tian, M.; Li, X.; Li, J.-J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Alexe, G.; Dharia, N.V.; Ross, L.; Iniguez, A.B.; Conway, A.S.; Wang, E.J.; Veschi, V.; Lam, N.; Qi, J.; et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Investig. 2017, 128, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, J.; Liu, T.; Atmadibrata, B.; Bradner, E.J.; Marshall, G.M.; Lock, R.B. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-Myc expression and induce anticancer effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hao, D.; Wang, L.; Wang, H.; Wang, Y.; Zhao, Z.; Li, P.; Deng, C.; Di, L.-J. Epigenetic targeting drugs potentiate chemotherapeutic effects in solid tumor therapy. Sci. Rep. 2017, 7, 4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raynal, N.J.-M.; Da Costa, E.M.; Lee, J.T.; Gharibyan, V.; Ahmed, S.; Zhang, H.; Sato, T.; Malouf, G.G.; Issa, J.-P.J. Repositioning FDA-Approved Drugs in Combination with Epigenetic Drugs to Reprogram Colon Cancer Epigenome. Mol. Cancer Ther. 2016, 16, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L Inhibitor EPZ-5676 Displays Synergistic Antiproliferative Activity in Combination with Standard of Care Drugs and Hypomethylating Agents in MLL-Rearranged Leukemia Cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Ramadoss, M.; Mahadevan, V. Targeting the cancer epigenome: Synergistic therapy with bromodomain inhibitors. Drug Discov. Today 2018, 23, 76–89. [Google Scholar] [CrossRef]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi-Borojeni, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef]
- Qin, W.; Zheng, Y.; Qian, B.; Zhao, M. Prostate Cancer Stem Cells and Nanotechnology: A Focus on Wnt Signaling. Front. Pharmacol. 2017, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chenna, V.; Hu, C.; Sun, H.-X.; Khan, M.; Bai, H.; Yang, X.-R.; Zhu, Q.-F.; Sun, Y.-F.; Maitra, A.; et al. Polymeric Nanoparticle Encapsulated Hedgehog Pathway Inhibitor HPI-1 (“NanoHHI”) Inhibits Systemic Metastases in an Orthotopic Model of Human Hepatocellular Carcinoma. Clin. Cancer Res. 2011, 18, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- Burke, A.R.; Singh, R.; Carroll, D.L.; Wood, J.C.S.; D’Agostino, R.B.; Ajayan, P.M.; Torti, F.M.; Torti, S.V. The resistance of breast cancer stem cells to conventional hyperthermia and their sensitivity to nanoparticle-mediated photothermal therapy. Biomaterials 2012, 33, 2961–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.-J.; Zhang, Y.; Sun, L.; Liu, Y. The effect of hyaluronic acid functionalized carbon nanotubes loaded with salinomycin on gastric cancer stem cells. Biomaterials 2014, 35, 9208–9223. [Google Scholar] [CrossRef] [PubMed]





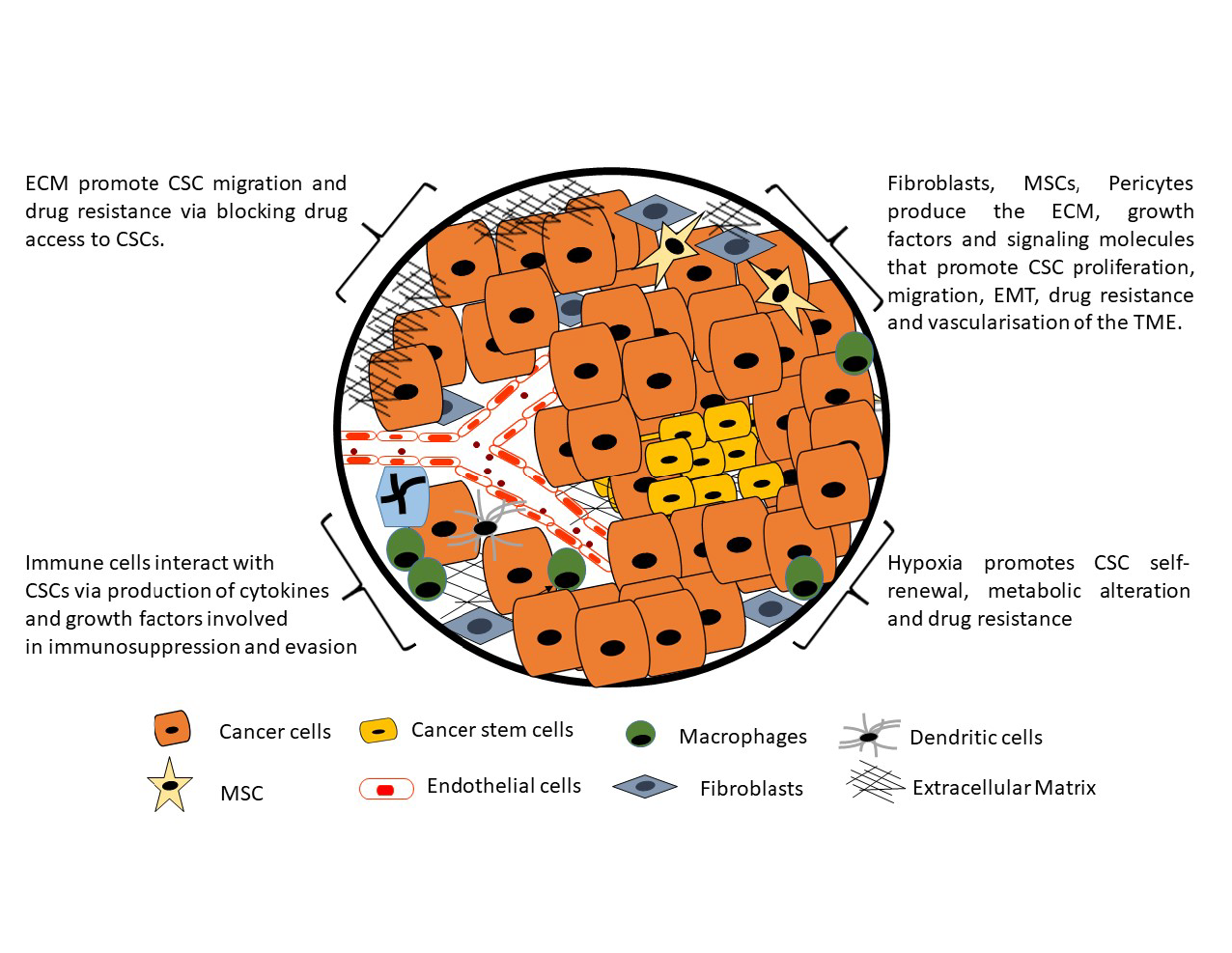{kind=link}
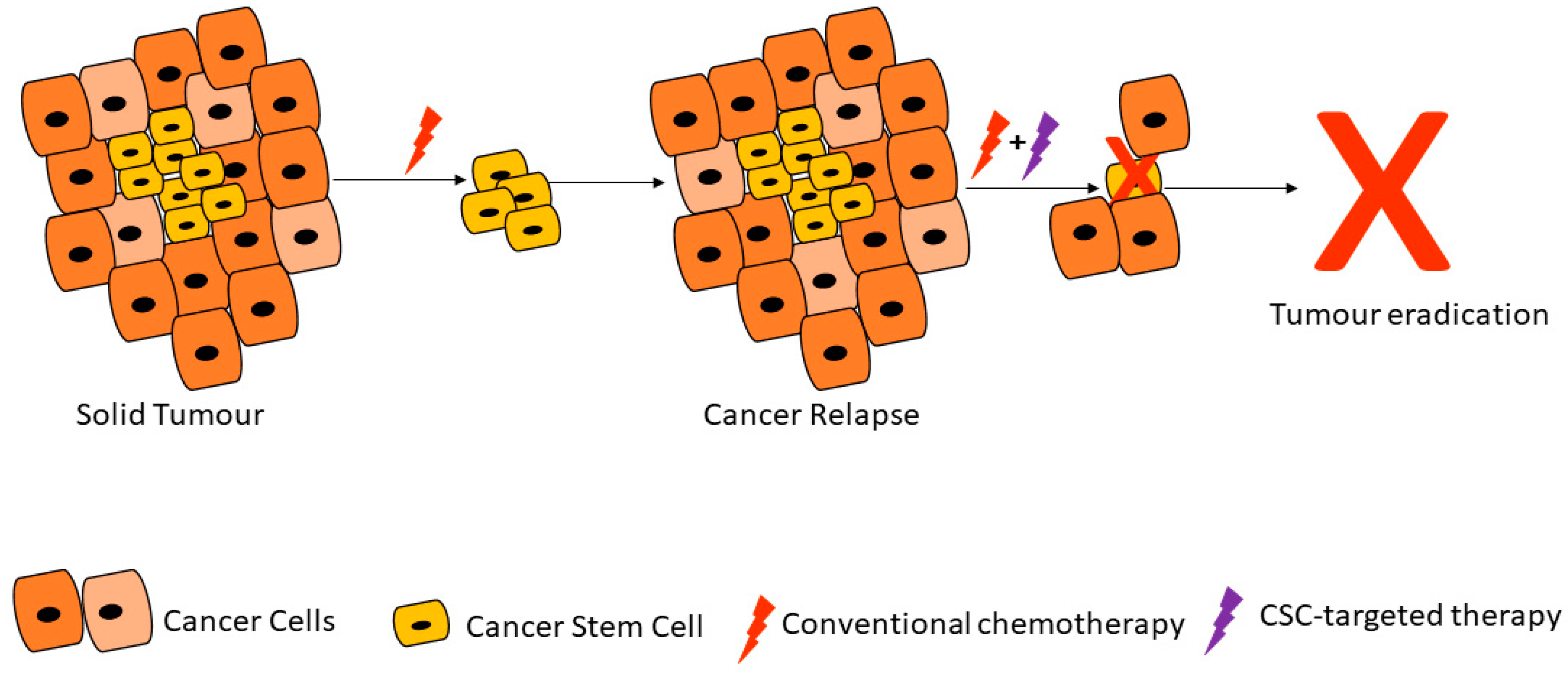{kind=link}
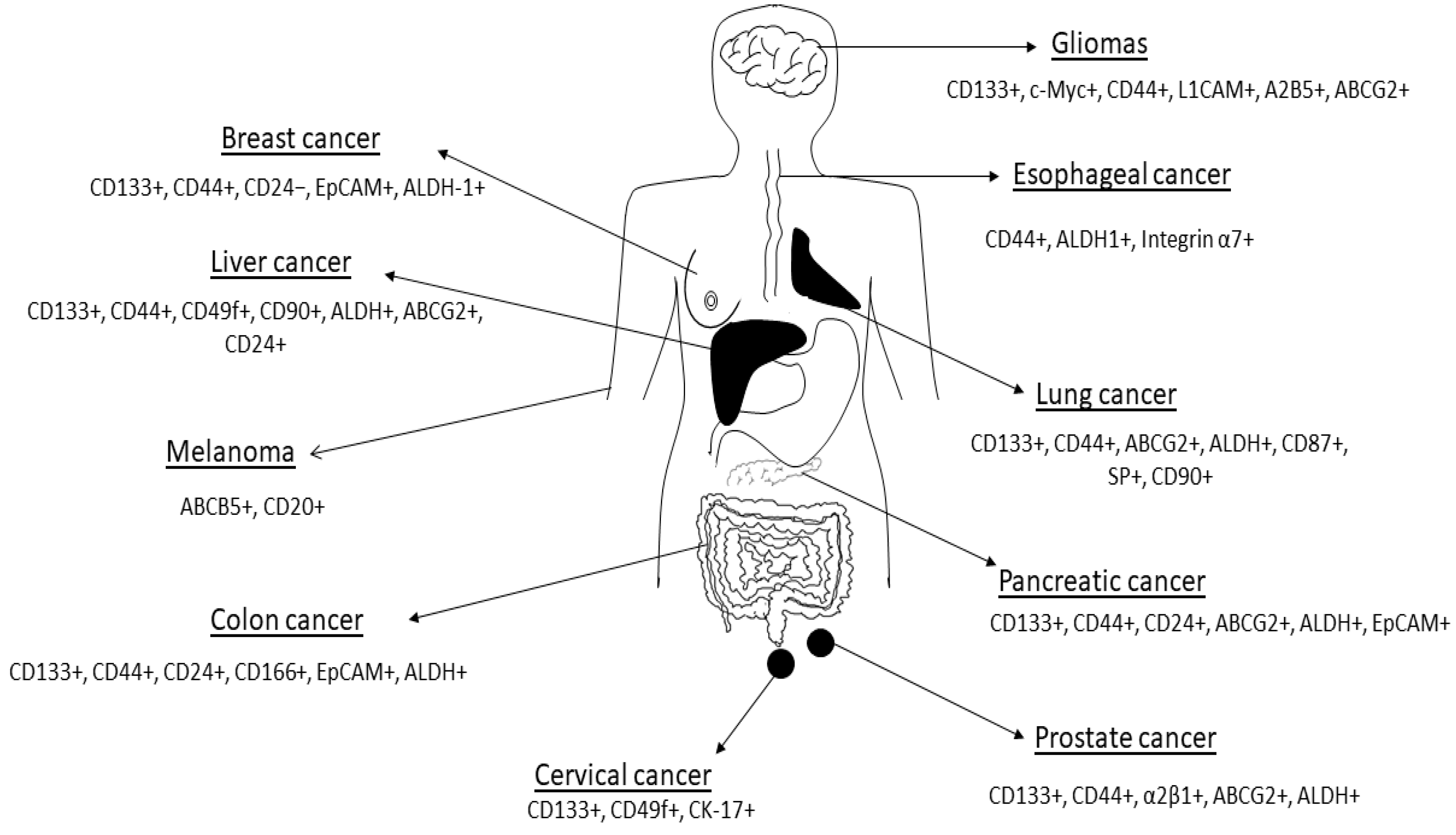{kind=link}
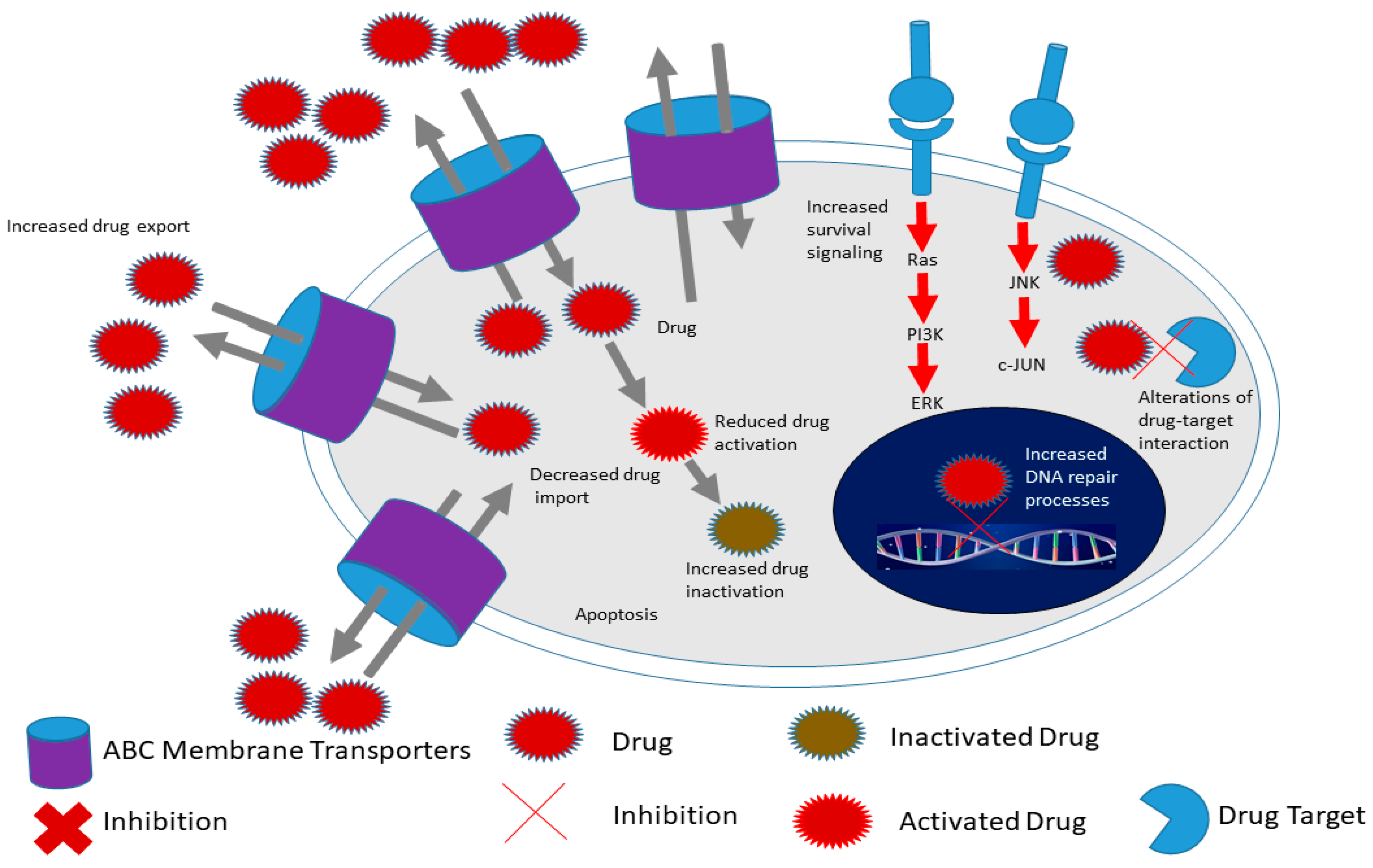{kind=link}
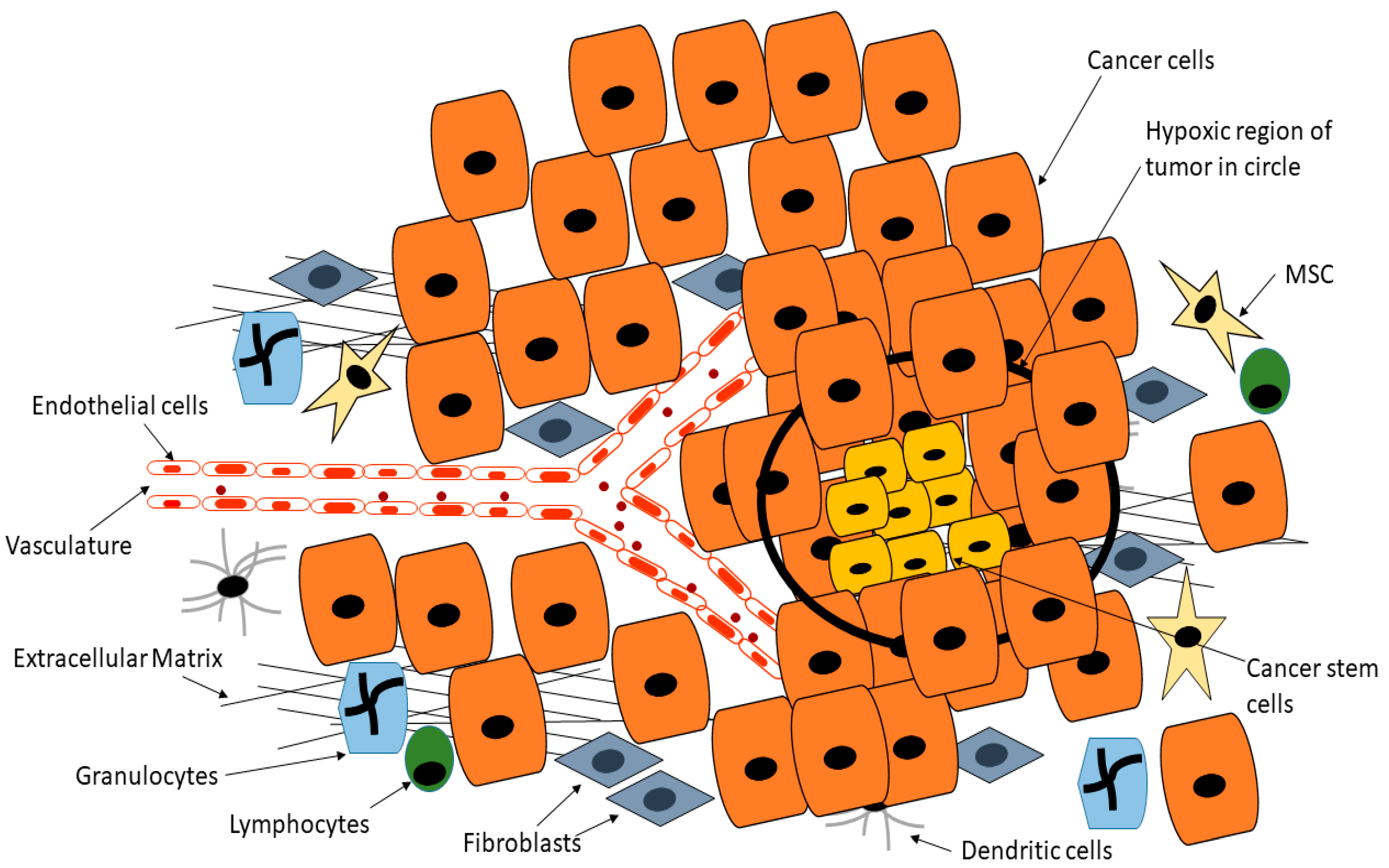{kind=link}
| Cancer | CSC Markers | References |
|---|---|---|
| Cervical | CD133+, CD49f+, CK-17+ | [35,36,37] |
| Esophageal | CD44+, ALDH1+, Integrin α7+ | [38,39] |
| Kidney | CD24-, CD44+, CD105+, CD133+ | [40,41,42] |
| Lung cancer | CD44+, CD90+, CD133+, ABCG2+, ALDH+ | [24,33] |
| Colon cancer | CD24+, CD44+, CD133+, EpCAM+, ALDH+ | [43,44,45,46] |
| Liver cancer | CD24+, CD44+, CD90+, CD133+, ALDH+, ABCG2+ | [47,48] |
| Breast cancer | CD24-, CD44+, CD133+, ALDH-1+ | [5,49,50] |
| Gastric | CD44+, CD133+ | [51,52,53] |
| Glioma | CD44+, CD133+, A2B5+, BCRP1+, SSEA-1+ | [54,55] |
| Leukemia (AML) | CD34+, CD38−, CD123+ | [56,57,58] |
| Leukemia (CML) | CD25+, CD26+, CD44+, CD93+, IL1RAP+ | [59,60] |
| Ovarian | CD44+, CD117+, CD133+, ALDH1+ | [61,62] |
| Prostate cancer | CD44+, CD133+, α2β1+, ALDH+ | [63,64,65] |
| Pancreatic cancer | CD44+, CD133+, ABCG2+, ALDH+, EpCAM+ | [66,67,68] |
| Melanoma | ABCB5+, CD20+ | [69,70] |
| Head and neck cancer | CD44+, CD133+ | [71,72] |
| Sarcoma | CD29+, CD117+, CD133+, Nestin+, Stro-1+ | [73,74] |
| Cancer Type | Chemotherapy/Radiotherapy/Immunotherapy | Clinical Trial Identifier |
|---|---|---|
| Breast | Ruxolitinib + Chemotherapy | NCT02876302 |
| Lapatinib + Radiotherapy | NCT01868503 | |
| Paclitaxel + Reparixin | NCT02370238 | |
| Paclitaxel + Reparixin | NCT02001974 | |
| Vorinostat + Lapatinib | NCT01118975 | |
| MK-0752 + Docetaxel + Pegfilgrastim | NCT00645333 | |
| Colorectal | OMP-305B83 + FOLFIRI + FOLFOX | NCT03035253 |
| Napabucasin + Fluorouracil + Leucovorin + Irinotecan + Bevacizumab | NCT02753127 | |
| OMP-21M18 | NCT01189942 | |
| Esophageal | Dietary Supplement: Fursultiamine | NCT02423811 |
| Gastrointestinal | Phase 1: BBI608 Phase 2: Fluorouracil + Oxaliplatin + Leucovorin + Irinotecan + Bevacizumab + Capecitabine + Regorafenib | NCT02024607 |
| Glioma | 3-Dimensional Conformal Radiation Therapy + Gamma-Secretase Inhibitor RO4929097 + Intensity-Modulated Radiation Therapy + Temozolomide | NCT01119599 |
| ChemoID assay + Chemotherapy | NCT03632135 | |
| Stem Cell Radiotherapy (ScRT) + Temozolomide | NCT02039778 | |
| Head and Neck | IPI-926 + Cetuximab | NCT01255800 |
| Hematologic | Azacitidine + SL-401 + Venetoclax | NCT03113643 |
| Lenalidomide + Dexamethasone + MEDI-551 | NCT01861340 | |
| Zileuton | NCT01130688 | |
| Hepatocellular | BBI608 + BBI503 + Sorafenib | NCT02279719 |
| Metformin | NCT01442870 | |
| Ovarian | Chemotherapy | NCT03632798 |
| Carboplatin + Paclitaxel + Ruxolitinib + Ruxolitinib Phosphate | NCT02713386 | |
| Metformin | NCT01579812 | |
| Pancreatic | Gamma-secretase/Notch signaling pathway inhibitor RO4929097 | NCT01192763 |
| Demcizumab + Abraxane® + Gemcitabine | NCT01189929 | |
| Cyberknife radiation + gemcitabine | NCT01051284 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896. https://doi.org/10.3390/cells9081896
Dzobo K, Senthebane DA, Ganz C, Thomford NE, Wonkam A, Dandara C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells. 2020; 9(8):1896. https://doi.org/10.3390/cells9081896
Chicago/Turabian StyleDzobo, Kevin, Dimakatso Alice Senthebane, Chelene Ganz, Nicholas Ekow Thomford, Ambroise Wonkam, and Collet Dandara. 2020. "Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review" Cells 9, no. 8: 1896. https://doi.org/10.3390/cells9081896
APA StyleDzobo, K., Senthebane, D. A., Ganz, C., Thomford, N. E., Wonkam, A., & Dandara, C. (2020). Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells, 9(8), 1896. https://doi.org/10.3390/cells9081896