TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Challenge Assays
2.2. siRNA Assay
2.3. Real-Time PCR
2.4. TNF and IL-8 Analysis by ELISA
2.5. Animal Model
2.6. Statistical Analysis
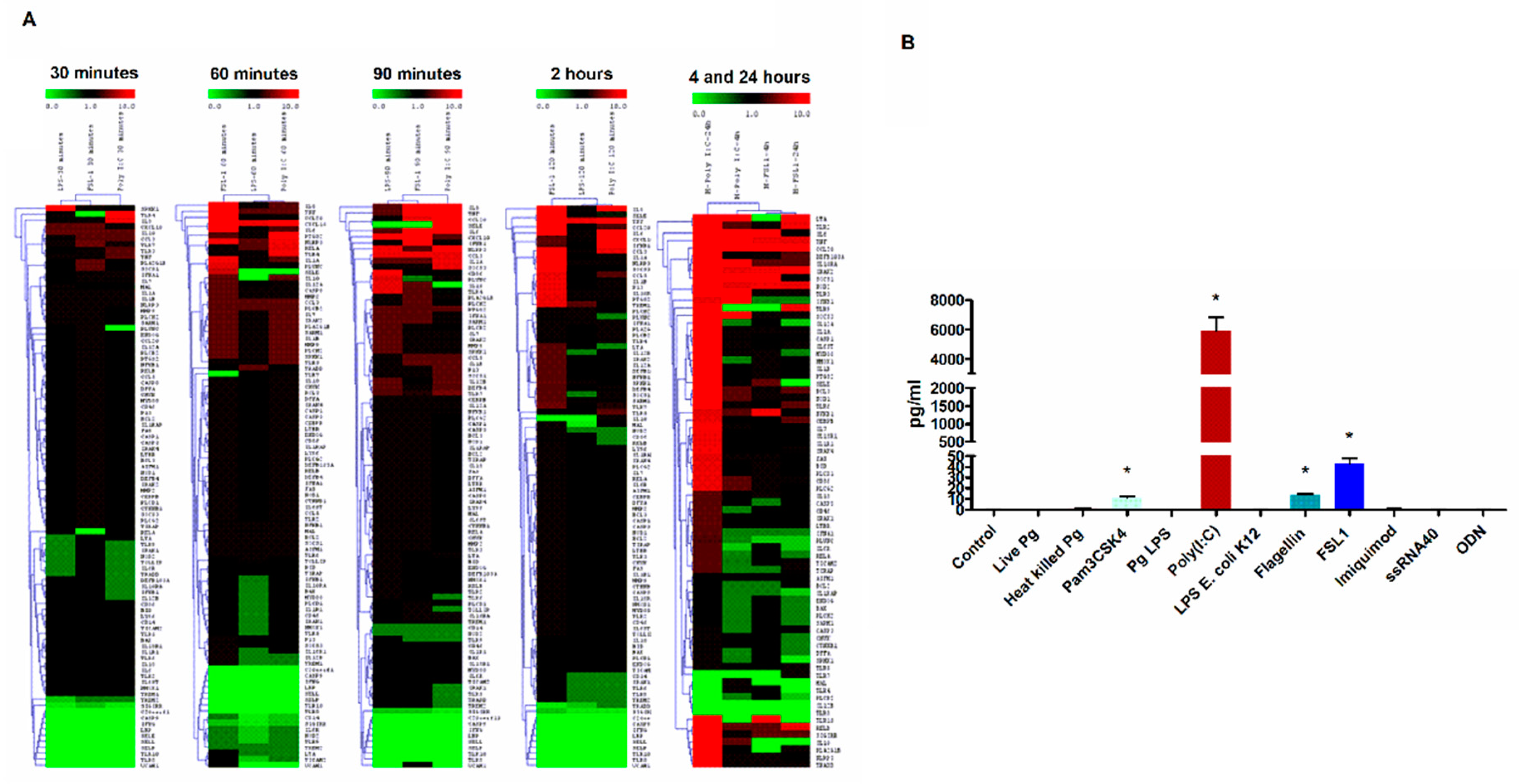
3. Results
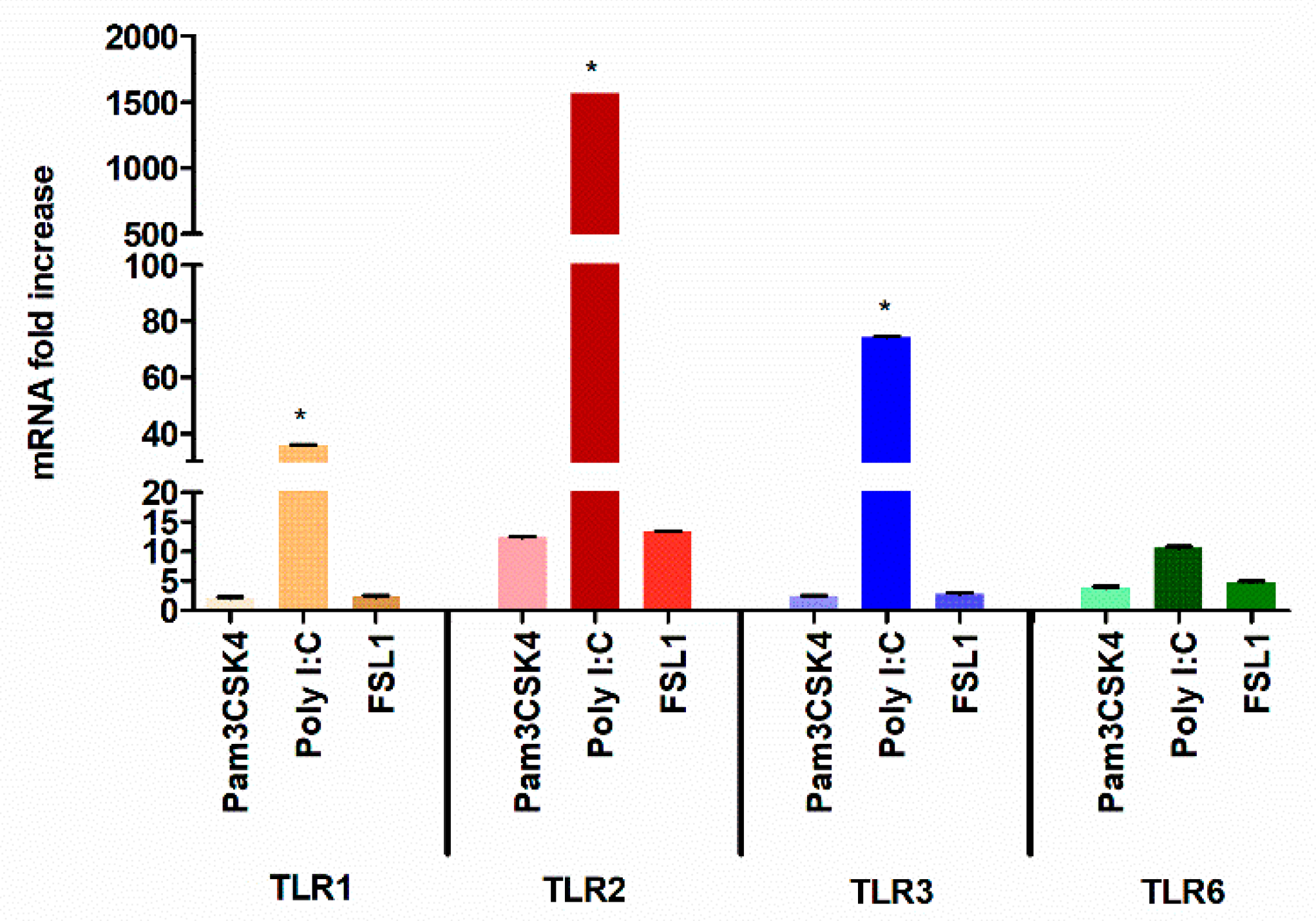
3.1. TLR3 Stimulation Leads to Robust TLR2 Transcriptional Activation
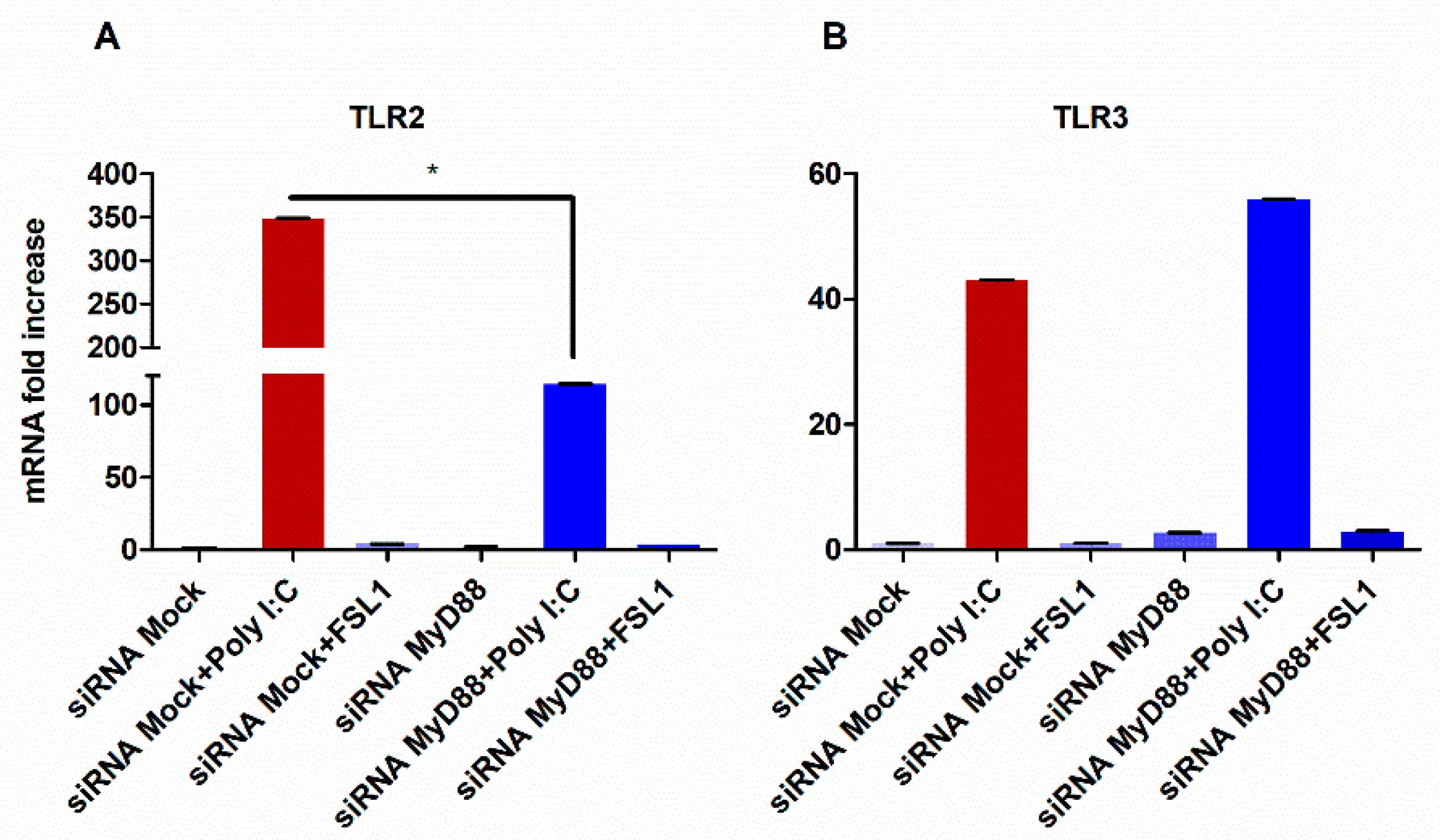
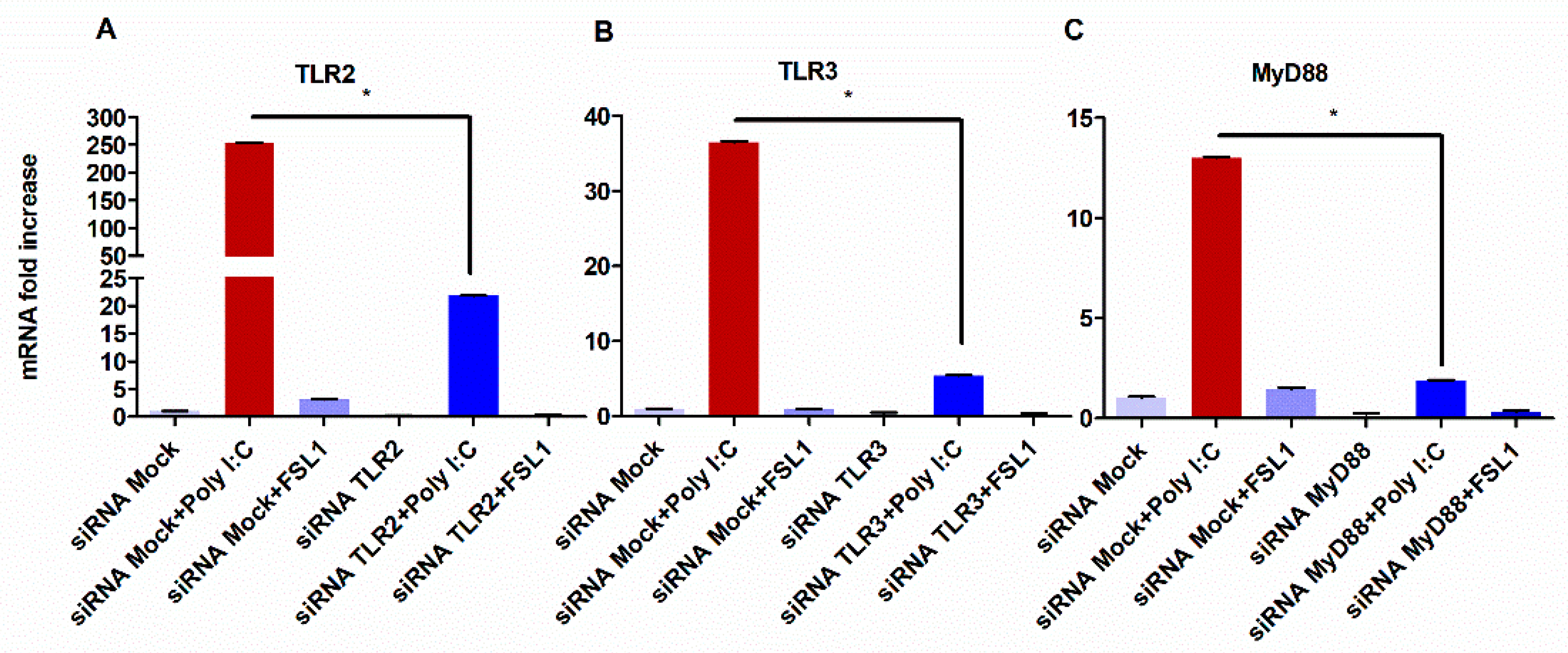
3.2. TLR3-Mediated Expression of TLR2 Requires MyD88 Signaling
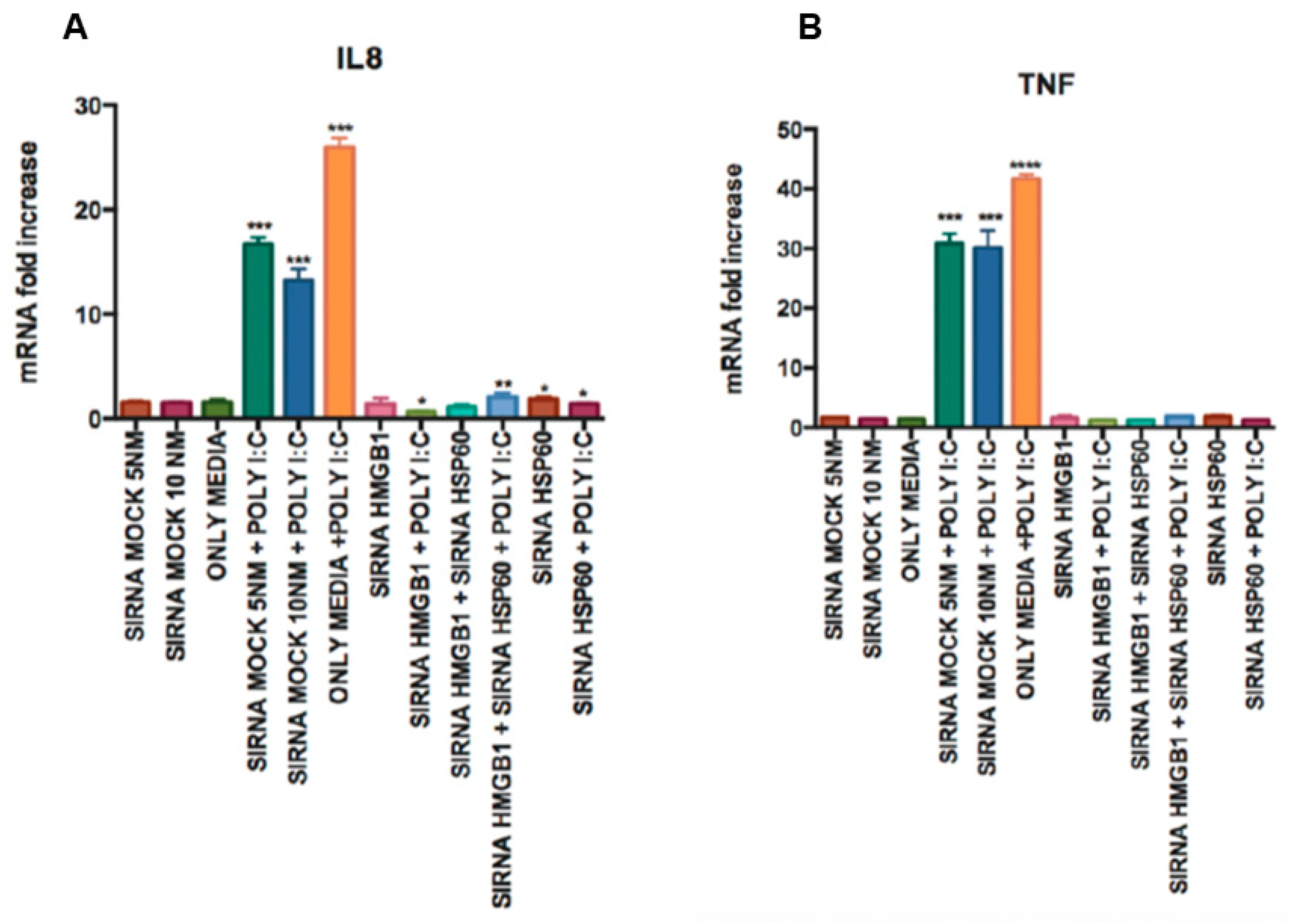
3.3. TLR3 Signaling Potentiate Pro-Inflammatory Cytokine Secretion Partially through the Activation of TLR2 and MyD88
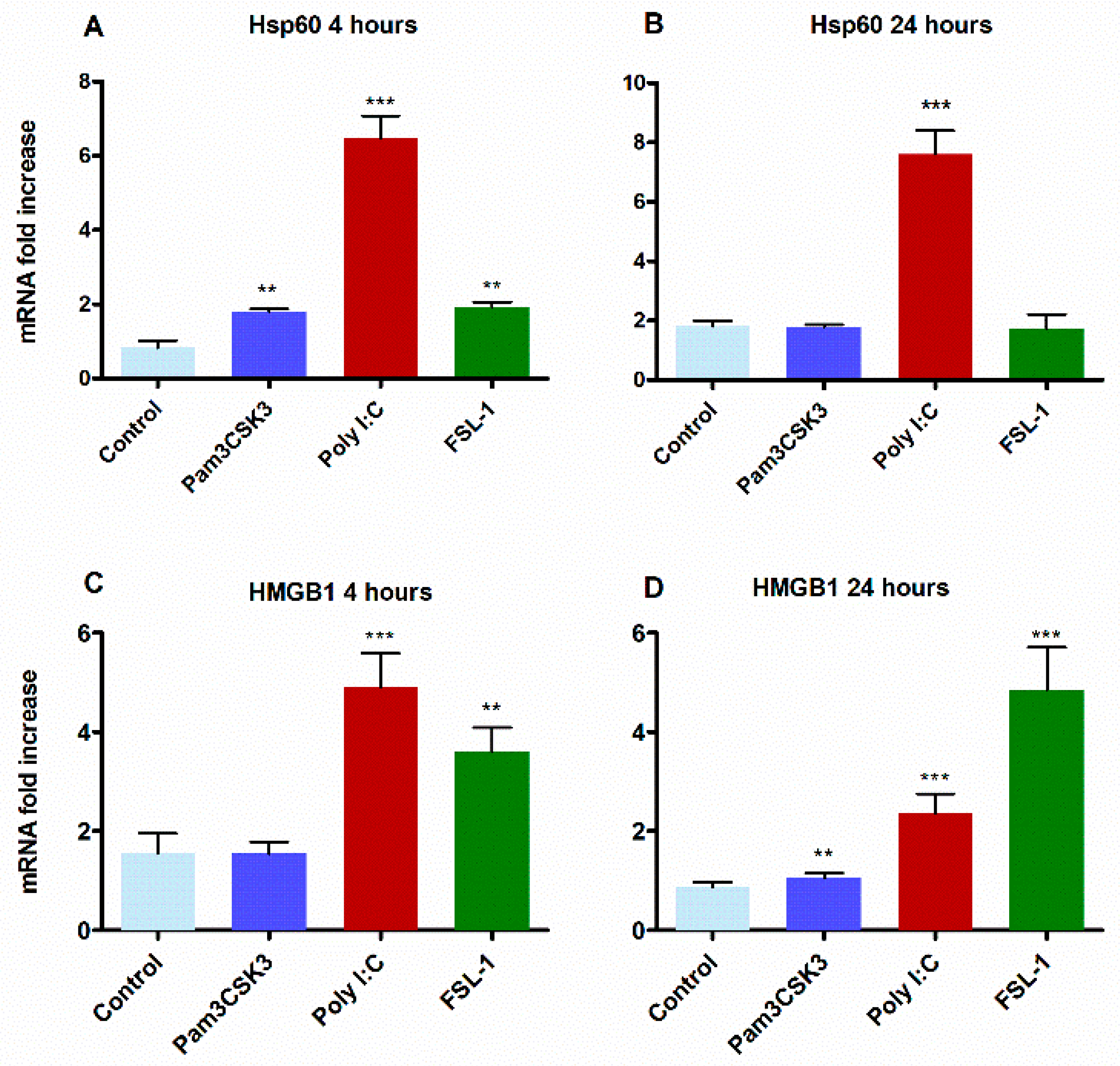
3.4. Activation of TLR2 by TLR3 is Mediated via the Induction of Endogenous Ligands HMGB1 and Hsp60
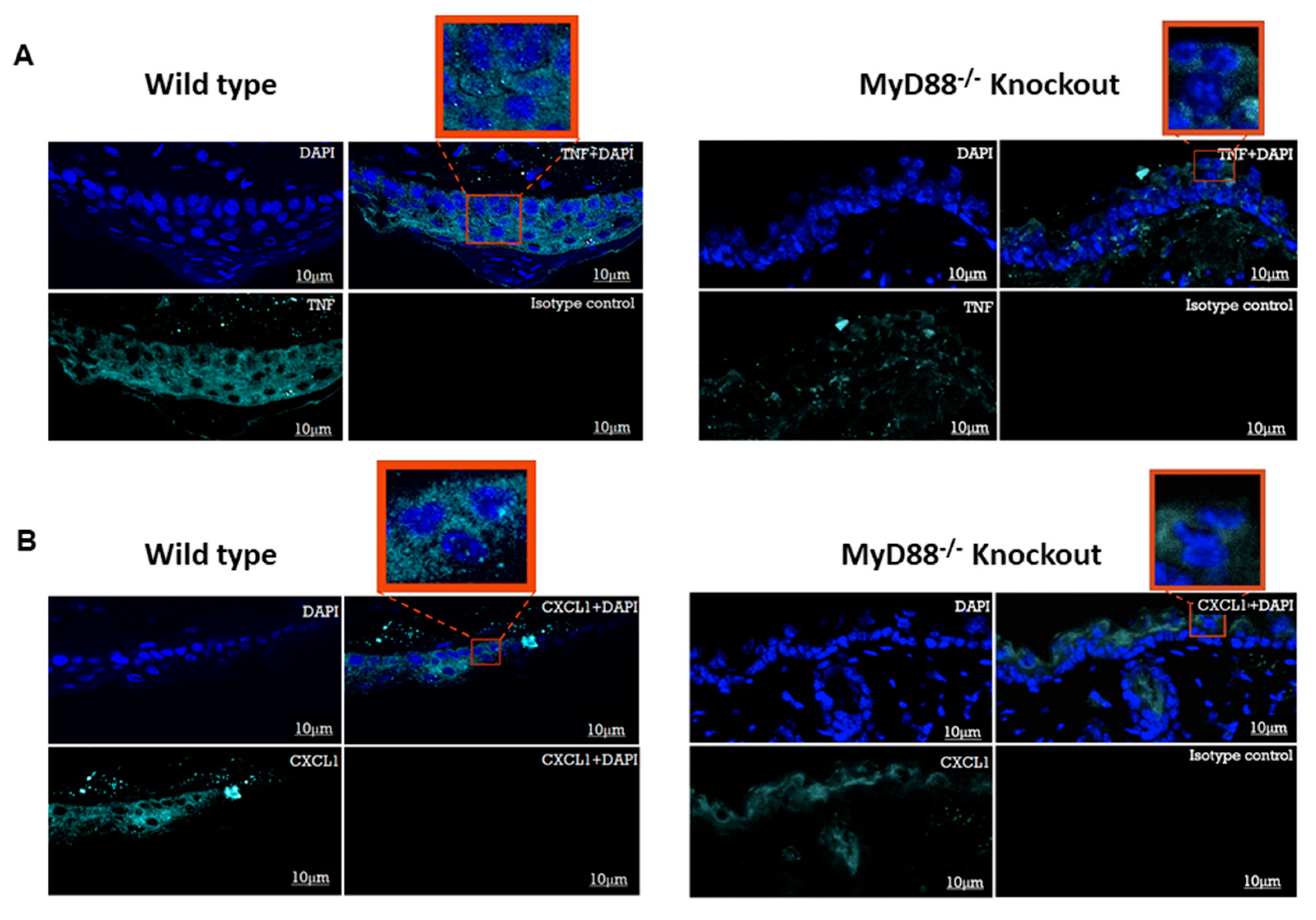
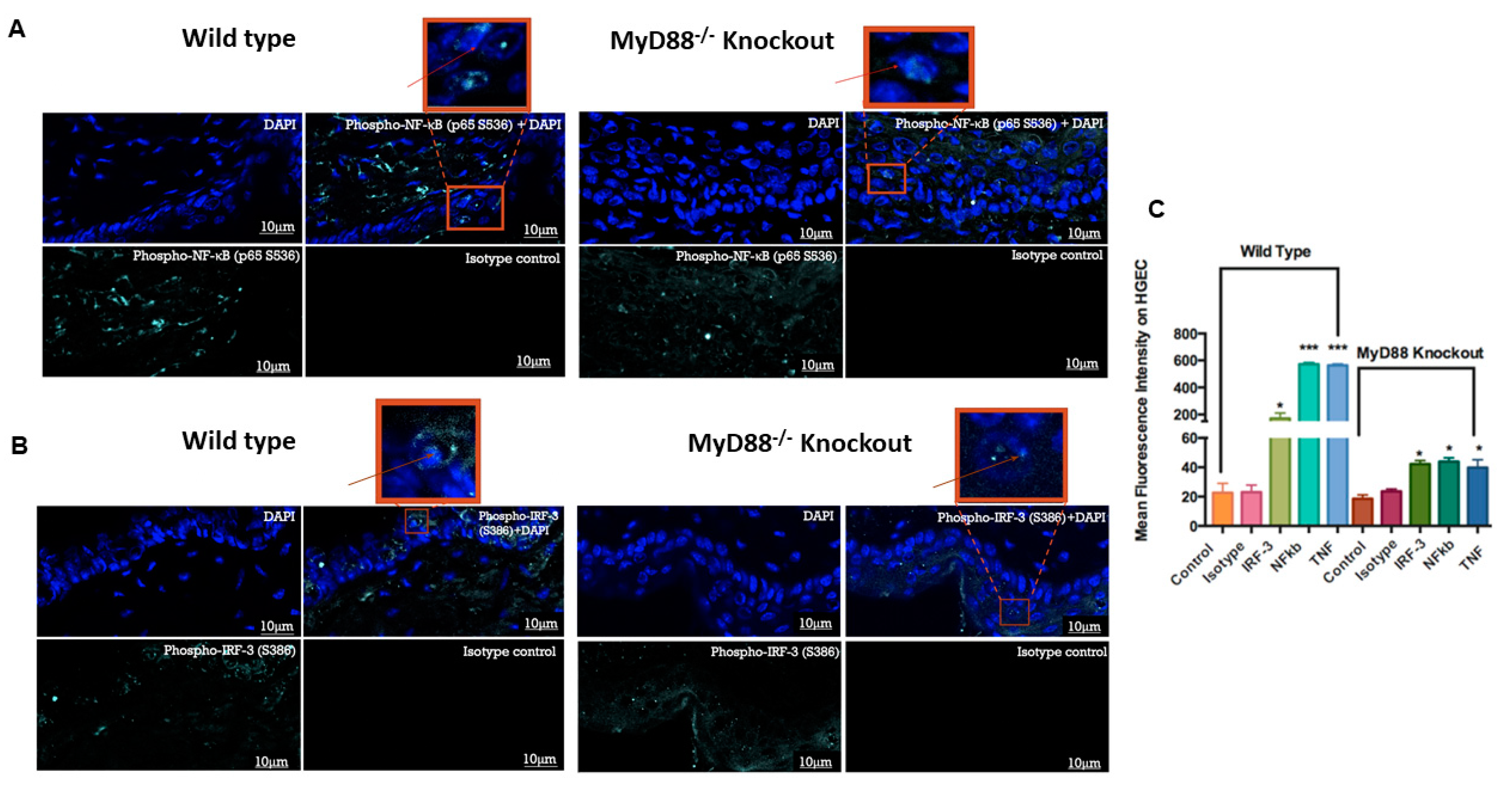
3.5. Myd88−/− Knockout Mouse Downregulated the Pro-Inflammatory Genes and Transcription Factors NF-κB and IRF3
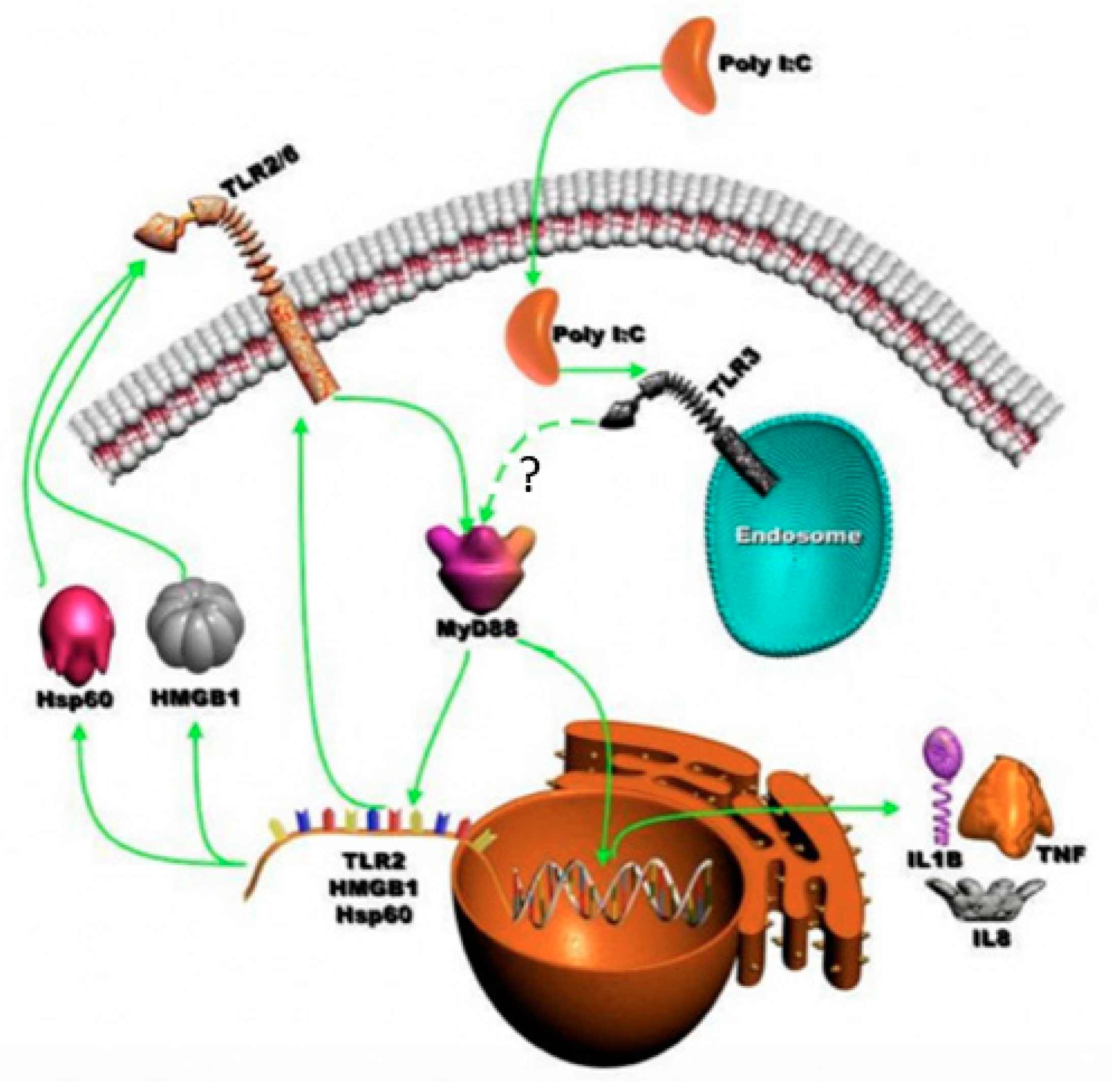
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Alkanani, A.K.; Hara, N.; Lien, E.; Ir, D.; Kotter, C.V.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Induction of diabetes in the RIP-B7.1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes 2014, 63, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beklen, A.; Sarp, A.S.; Uckan, D.; Tsaous Memet, G. The function of TLR4 in interferon gamma or interleukin-13 exposed and lipopolysaccharide stimulated gingival epithelial cell cultures. Biotech. Histochem. 2014, 89, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Mendoza, C.; Layunta, E.; Alcalde, A.I.; Mesonero, J.E. TLR2, TLR3, and TLR4 activation specifically alters the oxidative status of intestinal epithelial cells. Cell Stress Chaperones 2014, 19, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Tan, X.; Zhou, Q.; Tian, Y.; Kijlstra, A.; Yang, P. TLR3 and TLR4 But not TLR2 are involved in vogt-koyanagi- harada disease by triggering proinflammatory cytokines production through promoting the production of mitochondrial reactive oxygen species. Curr. Mol. Med. 2015, 15, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.; Barker, G.; Lappas, M. TLR2, TLR3 and TLR5 regulation of pro-inflammatory and pro-labour mediators in human primary myometrial cells. J. Reprod. Immunol. 2017, 122, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.; Dunne, A.; Brikos, C.; Jefferies, C.A.; Doyle, S.L.; LA, O.N. MyD88 adapter-like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signal transduction. J. Biol. Chem. 2016, 291, 26240. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Cong, X.; Chen, L.; Qi, J.; Wu, X.; Zhou, M.; Yoo, D.; Li, F.; Sun, W.; Wu, J.; et al. Synergy of TLR3 and 7 ligands significantly enhances function of DCs to present inactivated PRRSV antigen through TRIF/MyD88-NF-kappaB signaling pathway. Sci. Rep. 2016, 6, 23977. [Google Scholar] [CrossRef]
- Thorburn, A.N.; Tseng, H.Y.; Donovan, C.; Hansbro, N.G.; Jarnicki, A.G.; Foster, P.S.; Gibson, P.G.; Hansbro, P.M. TLR2, TLR4 AND MyD88 Mediate Allergic Airway Disease (AAD) and streptococcus pneumoniae-induced suppression of AAD. PLoS ONE 2016, 11, e0156402. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Benakanakere, M.; Kinane, D.F. Innate cellular responses to the periodontal biofilm. Front. Oral. Biol. 2012, 15, 41–55. [Google Scholar] [PubMed]
- Benakanakere, M.R.; Li, Q.; Eskan, M.A.; Singh, A.V.; Zhao, J.; Galicia, J.C.; Stathopoulou, P.; Knudsen, T.B.; Kinane, D.F. Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J. Biol. Chem. 2009, 284, 23107–23115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskan, M.A.; Benakanakere, M.R.; Rose, B.G.; Zhang, P.; Zhao, J.; Stathopoulou, P.; Fujioka, D.; Kinane, D.F. Interleukin-1beta modulates proinflammatory cytokine production in human epithelial cells. Infect. Immun. 2008, 76, 2080–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinane, D.F.; Galicia, J.C.; Gorr, S.U.; Stathopoulou, P.G.; Benakanakere, M.P. gingivalis interactions with epithelial cells. Front. Biosci. 2008, 13, 966–984. [Google Scholar] [CrossRef]
- Darveau, R.P.; Pham, T.T.; Lemley, K.; Reife, R.A.; Bainbridge, B.W.; Coats, S.R.; Howald, W.N.; Way, S.S.; Hajjar, A.M. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect. Immun. 2004, 72, 5041–5051. [Google Scholar] [CrossRef] [Green Version]
- Burns, E.; Bachrach, G.; Shapira, L.; Nussbaum, G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J. Immunol. 2006, 177, 8296–8300. [Google Scholar] [CrossRef] [Green Version]
- Bernard, Q.; Gallo, R.L.; Jaulhac, B.; Nakatsuji, T.; Luft, B.; Yang, X.; Boulanger, N. Ixodes tick saliva suppresses the keratinocyte cytokine response to TLR2/TLR3 ligands during early exposure to Lyme borreliosis. Exp. Dermatol. 2016, 25, 26–31. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Benavidez, J.M.; da Costa, A.; Mayfield, J.; Harris, R.A. Sedative and motor incoordination effects of ethanol in mice lacking CD14, TLR2, TLR4, or MyD88. Alcohol. Clin. Exp. Res. 2017, 41, 531–540. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Chernis, J.; da Costa, A.; Mayfield, J.; Harris, R.A. Ethanol consumption in mice lacking CD14, TLR2, TLR4, or MyD88. Alcohol. Clin. Exp. Res. 2017, 41, 516–530. [Google Scholar] [CrossRef]
- Cao, L.; Ge, X.; Gao, Y.; Ren, Y.; Ren, X.; Li, G. Porcine epidemic diarrhea virus infection induces NF-kappaB activation through the TLR2, TLR3 and TLR9 pathways in porcine intestinal epithelial cells. J. Gen. Virol. 2015, 96, 1757–1767. [Google Scholar] [CrossRef]
- Chen, D.; Xie, W.; Lu, Y.; Su, S.; Nong, L.; Jia, Y.; Liu, Y.; Zhou, W.; Wang, H.; Tan, A. Gene polymorphisms of TLR2 and TLR3 in HBV clearance and HBV-related hepatocellular carcinoma in a Chinese male population. Int. J. Biol. Markers 2017, 32, e195–e201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Su, L.; Xu, Q.; Katz, J.; Michalek, S.M.; Fan, M.; Feng, X.; Zhang, P. IL-1R/TLR2 through MyD88 divergently modulates osteoclastogenesis through regulation of Nuclear Factor of Activated T Cells c1 (NFATc1) and B Lymphocyte-induced Maturation Protein-1 (Blimp1). J. Biol. Chem. 2015, 290, 30163–30174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Gao, Y.; Mu, D.G.; Fu, E.Q. MiR-23a-5p modulates mycobacterial survival and autophagy during mycobacterium tuberculosis infection through TLR2/MyD88/NF-kappaB pathway by targeting TLR2. Exp. Cell Res. 2017, 354, 71–77. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Yoshida, Y.; Arashidani, K.; Yoshida, S.; Takano, H.; Sun, G.; Shibamoto, T. Urban PM2.5 exacerbates allergic inflammation in the murine lung via a TLR2/TLR4/MyD88-signaling pathway. Sci. Rep. 2017, 7, 11027. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, C.L.; Najdovska, M.; Tye, H.; McLeod, L.; Yu, L.; Jarnicki, A.; Bhathal, P.S.; Putoczki, T.; Ernst, M.; Jenkins, B.J. Differential role of MyD88 and Mal/TIRAP in TLR2-mediated gastric tumourigenesis. Oncogene 2014, 33, 2540–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Benakanakere, M.R.; Hosur, K.B.; Galicia, J.C.; Martin, M.; Kinane, D.F. Mammalian target of rapamycin (mTOR) regulates TLR3 induced cytokines in human oral keratinocytes. Mol. Immunol. 2010, 48, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.R.; Hooper, L.V.; Schmidt, R.R.; von Aulock, S.; et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef]
- Lakpour, M.R.; Koruji, M.; Shahverdi, A.; Aghajanpour, S.; Rajabian Naghandar, M.; Sadighi Gilani, M.A.; Sabbaghian, M.; Aflatoonian, R. The expression of TLR2 and TLR3 in sertoli cells of azoospermic patients. Cell J. 2017, 19, 375–385. [Google Scholar]
- Liang, S.; Xinyong, C.; Hongmin, Z.; Jing, W.; Lang, H.; Ping, Z. TLR2 and TLR3 expression as a biomarker for the risk of doxorubicin-induced heart failure. Toxicol. Lett. 2018, 295, 205–211. [Google Scholar] [CrossRef]
- Makni, L.; Messadi, A.; Zidi, S.; Gazouani, E.; Mezlini, A.; Yacoubi-Loueslati, B. TLR2 (-196 to -174 Ins/Del) and TLR3 (1377C>T) as biomarkers for nasopharyngeal cancer in Tunisia. Turk. J. Med. Sci. 2017, 47, 1216–1222. [Google Scholar] [CrossRef]
- Mortazavi, S.H.; Amin, R.; Alyasin, S.; Kashef, S.; Karimi, M.H.; Babaei, M.; Younesi, V. Down-regulation of TLR2, 3, 9 and signaling mediators, MyD88 and TRIF, gene transcript levels in patients with Kawasaki disease treated with IVIG. Iran. J. Allergy Asthma Immunol. 2015, 14, 188–197. [Google Scholar] [PubMed]
- Muralidharan, S.; Lim, A.; Catalano, D.; Mandrekar, P. Human binge alcohol intake inhibits TLR4-MyD88 and TLR4-TRIF responses but not the TLR3-TRIF pathway: HspA1A and PP1 play selective regulatory roles. J. Immunol. 2018, 200, 2291–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajalakshmy, A.R.; Malathi, J.; Madhavan, H.N. Hepatitis C virus NS3 mediated microglial inflammation via TLR2/TLR6 MyD88/NF-kappaB pathway and toll like receptor ligand treatment furnished immune tolerance. PLoS ONE 2015, 10, e0125419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramstead, A.G.; Robison, A.; Blackwell, A.; Jerome, M.; Freedman, B.; Lubick, K.J.; Hedges, J.F.; Jutila, M.A. Roles of Toll-Like Receptor 2 (TLR2), TLR4, and MyD88 during pulmonary coxiella burnetii infection. Infect. Immun. 2016, 84, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, N.; Mirahmadian, M.; Jeddi-Tehrani, M.; Rezania, S.; Ghasemi, J.; Kazemnejad, S.; Mirzadegan, E.; Vafaei, S.; Kashanian, M.; Rasoulzadeh, Z.; et al. Lipopolysaccharide- and lipoteichoic acid-mediated pro-inflammatory cytokine production and modulation of TLR2, TLR4 and MyD88 expression in human endometrial cells. J. Reprod. Infertil. 2015, 16, 72–81. [Google Scholar]
- Ren, X.; Wang, L.; Wu, X. A potential link between TSLP/TSLPR/STAT5 and TLR2/MyD88/NFkappaB-p65 in human corneal epithelial cells for Aspergillus fumigatus tolerance. Mol. Immunol. 2016, 71, 98–106. [Google Scholar] [CrossRef]
- Sahoo, B.R.; Basu, M.; Swain, B.; Dikhit, M.R.; Jayasankar, P.; Samanta, M. Elucidation of novel structural scaffold in rohu TLR2 and its binding site analysis with peptidoglycan, lipoteichoic acid and zymosan ligands, and downstream MyD88 adaptor protein. BioMed Res. Int. 2013, 2013, 185282. [Google Scholar] [CrossRef]
- Scheeren, F.A.; Kuo, A.H.; van Weele, L.J.; Cai, S.; Glykofridis, I.; Sikandar, S.S.; Zabala, M.; Qian, D.; Lam, J.S.; Johnston, D.; et al. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat. Cell Biol. 2014, 16, 1238–1248. [Google Scholar] [CrossRef]
- Singh, B.; Biswas, I.; Bhagat, S.; Surya Kumari, S.; Khan, G.A. HMGB1 facilitates hypoxia-induced vWF upregulation through TLR2-MYD88-SP1 pathway. Eur. J. Immunol. 2016, 46, 2388–2400. [Google Scholar] [CrossRef] [Green Version]
- Takehara, M.; Seike, S.; Takagishi, T.; Kobayashi, K.; Nagahama, M. Peptidoglycan accelerates granulopoiesis through a TLR2- and MyD88-dependent pathway. Biochem. Biophys. Res. Commun. 2017, 487, 419–425. [Google Scholar] [CrossRef]
- Talreja, D.; Singh, P.K.; Kumar, A. In vivo role of TLR2 and MyD88 signaling in eliciting innate immune responses in staphylococcal endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1719–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benakanakere, M.; Abdolhosseini, M.; Hosur, K.; Finoti, L.S.; Kinane, D.F. TLR2 promoter hypermethylation creates innate immune dysbiosis. J. Dent. Res. 2015, 94, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulou, P.G.; Benakanakere, M.R.; Galicia, J.C.; Kinane, D.F. The host cytokine response to Porphyromonas gingivalis is modified by gingipains. Oral. Microbiol. Immunol. 2009, 24, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopoulou, P.G.; Benakanakere, M.R.; Galicia, J.C.; Kinane, D.F. Epithelial cell pro-inflammatory cytokine response differs across dental plaque bacterial species. J. Clin. Periodontol. 2010, 37, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Benakanakere, M.R.; Zhao, J.; Galicia, J.C.; Martin, M.; Kinane, D.F. Sphingosine Kinase-1 is required for toll mediated β-Defensin 2 induction in human oral keratinocytes. PLoS ONE 2010, 5, e11512. [Google Scholar] [CrossRef] [Green Version]
- Kinane, D.F.; Shiba, H.; Stathopoulou, P.G.; Zhao, H.; Lappin, D.F.; Singh, A.; Eskan, M.A.; Beckers, S.; Waigel, S.; Alpert, B.; et al. Gingival epithelial cells heterozygous for Toll-like receptor 4 polymorphisms Asp299Gly and Thr399ile are hypo-responsive to Porphyromonas gingivalis. Genes Immun. 2006, 7, 190–200. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sakai, K.; Sanders, K.M.; Youssef, M.R.; Yanushefski, K.M.; Jensen, L.; Yosipovitch, G.; Akiyama, T. Mouse model of imiquimod-induced psoriatic itch. Pain 2016, 157, 2536–2543. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Z.; Jiang, L.; Chen, F.; Jin, R.; Cheng, L. Effects of oral and subcutaneous administration of HSP60 on myeloid-derived suppressor cells and atherosclerosis in ApoE-/- mice. Biochem. Biophys. Res. Commun. 2018, 498, 701–706. [Google Scholar] [CrossRef]
- Xia, J.; Winkelmann, E.R.; Gorder, S.R.; Mason, P.W.; Milligan, G.N. TLR3- and MyD88-dependent signaling differentially influences the development of West Nile virus-specific B cell responses in mice following immunization with RepliVAX WN, a single-cycle flavivirus vaccine candidate. J. Virol. 2013, 87, 12090–12101. [Google Scholar] [CrossRef] [Green Version]
- Szaleczky, E.; Pronai, L.; Molnar, B.; Berczi, L.; Feher, J.; Tulassay, Z. Increased cell proliferation in chronic Helicobacter pylori positive gastritis and gastric carcinoma--correlation between immuno-histochemistry and Tv image cytometry. Anal. Cell Pathol. 2000, 20, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barge, A.M.; Benajam, A.; Poirier, J.C.; Querne, G. Image analysis application at immuno-hematology (author’s transl). Pathol. Biol. 1979, 27, 235–240. [Google Scholar]
- Niflioglu, G.G.; Lebe, B. Pemphigoid gestationis: Light microscopic and direct immunofluorescense findings. Turk. Patoloji Derg. 2014, 30, 152–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Barranco, D.; Sandoval-Islas, M.E.; Trujillo-Valdes, V.M. Indirect immunofluorescense reaction in cysticercosis. Archivos de Investigacion Medica 1978, 9, 51–58. [Google Scholar]
- Kinane, J.A.; Benakanakere, M.R.; Zhao, J.; Hosur, K.B.; Kinane, D.F. Porphyromonas gingivalis influences actin degradation within epithelial cells during invasion and apoptosis. Cell Microbiol. 2012, 14, 1085–1096. [Google Scholar] [CrossRef]
- Eskan, M.A.; Rose, B.G.; Benakanakere, M.R.; Lee, M.J.; Kinane, D.F. Sphingosine 1-phosphate 1 and TLR4 mediate IFN-beta expression in human gingival epithelial cells. J. Immunol. 2008, 180, 1818–1825. [Google Scholar] [CrossRef]
- Eskan, M.A.; Rose, B.G.; Benakanakere, M.R.; Zeng, Q.; Fujioka, D.; Martin, M.H.; Lee, M.J.; Kinane, D.F. TLR4 and S1P receptors cooperate to enhance inflammatory cytokine production in human gingival epithelial cells. Eur. J. Immunol. 2008, 38, 1138–1147. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Perkins, D.J.; Vogel, S.N. Space and time: New considerations about the relationship between Toll-like receptors (TLRs) and type I interferons (IFNs). Cytokine 2015, 74, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, S.; Akira, S. Toll-like receptors and Type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.H.; Brunn, G.J.; Cascalho, M.; Platt, J.L. Pivotal advance: Endogenous pathway to SIRS, sepsis, and related conditions. J. Leukoc. Biol. 2007, 82, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leifer, C.A.; Medvedev, A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Talebi, S.; Anvar, A. Endogenous and exogenous natural adjuvants for vaccine development. Mini Rev. Med. Chem. 2017, 17, 1442–1456. [Google Scholar] [CrossRef] [PubMed]
- Shirjang, S.; Mansoori, B.; Solali, S.; Hagh, M.F.; Shamsasenjan, K. Toll-like receptors as a key regulator of mesenchymal stem cell function: An up-to-date review. Cell. Immunol. 2017, 315, 1–10. [Google Scholar] [CrossRef]
- Holzinger, D.; Foell, D.; Kessel, C. The role of S100 proteins in the pathogenesis and monitoring of autoinflammatory diseases. Mol. Cell Pediatr. 2018, 5, 7. [Google Scholar] [CrossRef]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.F., 3rd. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 2001, 276, 10229–10233. [Google Scholar] [CrossRef] [Green Version]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef]
- Miyake, K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 2007, 19, 3–10. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef]
- Zhang, P.; Cox, C.J.; Alvarez, K.M.; Cunningham, M.W. Cutting edge: Cardiac myosin activates innate immune responses through TLRs. J. Immunol. 2009, 183, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ofi, E.A.; Al-Ghamdi, B.S. High-Mobility Group Box 1 (HMGB1), an endogenous ligand of toll-like receptors 2 and 4, Induces astroglial inflammation via NF-beta pathway. Folia Morphol. 2018, 78, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, S.A.; Eidelman, A.S.; Ewer, J.C.; Ricca, J.M.; Serrano, A.; Tucker, K.C.; Vail, C.M.; Kurt, R.A. A role for HMGB1, HSP60 and Myd88 in growth of murine mammary carcinoma in vitro. Cell. Immunol. 2013, 282, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.; Herrup, E.A.; Warren, H.S.; Trigilio, J.; Shin, H.S.; Valentine, C.; Hellman, J. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J. Immunol. 2007, 178, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.F.; Talbot, S.; Gong, M.; Golenbock, D.T.; Bryant, C.E.; O’Neill, L.A. MyD88 adaptor-like is not essential for TLR2 signaling and inhibits signaling by TLR3. J. Immunol. 2009, 183, 3642–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Medzhitov, R. Control of adaptive immune responses by Toll-like receptors. Curr. Opin. Immunol. 2002, 14, 380–383. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.K.; Fisher, C.; Li, J.D.; Jiang, Y.; Huang, S.; Chen, L.Y. Bacterial LPS up-regulated TLR3 expression is critical for antiviral response in human monocytes: Evidence for negative regulation by CYLD. Int. Immunol. 2011, 23, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Bakaysa, S.L.; Potter, J.A.; Hoang, M.; Han, C.S.; Guller, S.; Norwitz, E.R.; Abrahams, V.M. Single- and double-stranded viral RNA generate distinct cytokine and antiviral responses in human fetal membranes. Mol. Hum. Reprod. 2014, 20, 701–708. [Google Scholar] [CrossRef]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Wissler, H.L.; Ehlerding, E.B.; Lyu, Z.; Zhao, Y.; Zhang, S.; Eshraghi, A.; Buuh, Z.Y.; McGuth, J.C.; Guan, Y.; Engle, J.W.; et al. Site-specific immuno-PET tracer to image PD-L1. Mol. Pharm. 2019, 16, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Shi, H.Y.; Wu, X.M.; Zhang, Y.H.; Zhong, Y.J.; Deng, W.J.; Zhang, Y.P.; Xie, M. Targeted inhibition of GATA-6 attenuates airway inflammation and remodeling by regulating caveolin-1 through TLR2/MyD88/NF-kappaB in murine model of asthma. Mol. Immunol. 2016, 75, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Granick, J.L.; Falahee, P.C.; Dahmubed, D.; Borjesson, D.L.; Miller, L.S.; Simon, S.I. Staphylococcus aureus recognition by hematopoietic stem and progenitor cells via TLR2/MyD88/PGE2 stimulates granulopoiesis in wounds. Blood 2013, 122, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Martinus, R.D.; Goldsbury, J. Endothelial TNF-alpha induction by Hsp60 secreted from THP-1 monocytes exposed to hyperglycaemic conditions. Cell Stress Chaperones 2018, 23, 519–525. [Google Scholar] [CrossRef]
- Mosaddad, S.A.; Tahmasebi, E.; Yazdanian, A.; Rezvani, M.B.; Seifalian, A.; Yazdanian, M.; Tebyanian, H. Oral microbial biofilms: An update. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 2005–2019. [Google Scholar] [CrossRef]
- Grinde, B.; Olsen, I. The role of viruses in oral disease. J. Oral Microbiol. 2010, 2, 2127. [Google Scholar] [CrossRef]
- Siqueira, J.F., Jr.; Rocas, I.N. The oral microbiota in health and disease: An overview of molecular findings. Methods Mol. Biol. 2017, 1537, 127–138. [Google Scholar]
- Teixeira, H.; Zhao, J.; Kinane, D.F.; Benakanakere, M.R. IFN-β secretion is through TLR3 but not TLR4 in human gingival epithelial cells. Mol. Immunol. 2019, 111, 27–31. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, H.S.; Zhao, J.; Kazmierski, E.; Kinane, D.F.; Benakanakere, M.R. TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response. Cells 2020, 9, 1910. https://doi.org/10.3390/cells9081910
Teixeira HS, Zhao J, Kazmierski E, Kinane DF, Benakanakere MR. TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response. Cells. 2020; 9(8):1910. https://doi.org/10.3390/cells9081910
Chicago/Turabian StyleTeixeira, Hellen S., Jiawei Zhao, Ethan Kazmierski, Denis F. Kinane, and Manjunatha R. Benakanakere. 2020. "TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response" Cells 9, no. 8: 1910. https://doi.org/10.3390/cells9081910
APA StyleTeixeira, H. S., Zhao, J., Kazmierski, E., Kinane, D. F., & Benakanakere, M. R. (2020). TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response. Cells, 9(8), 1910. https://doi.org/10.3390/cells9081910