Identification of Carbohydrate Metabolism Genes in the Metagenome of a Marine Biofilm Community Shown to Be Dominated by Gammaproteobacteria and Bacteroidetes

Abstract
:
1. Introduction
2. Results, Discussion
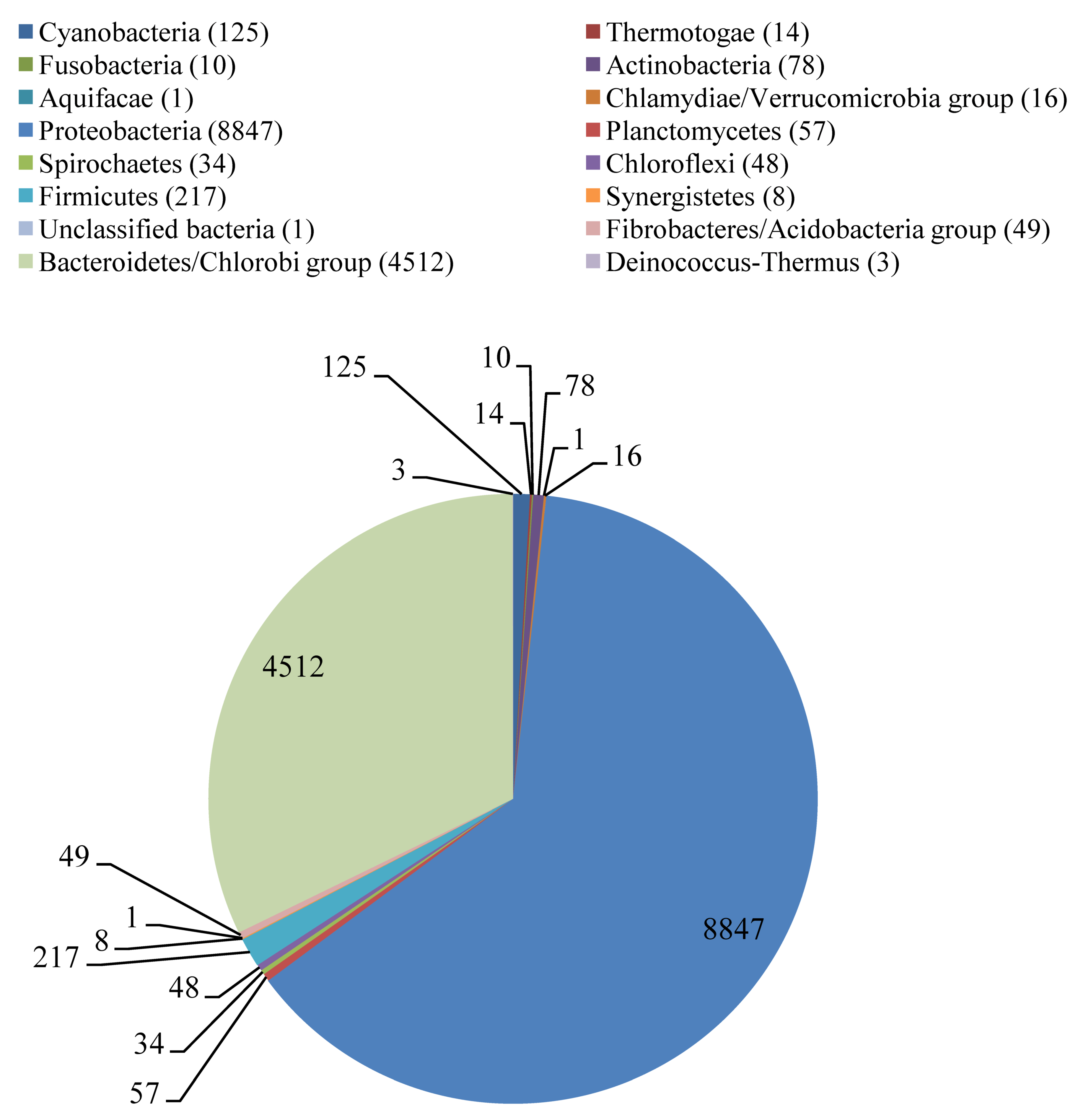
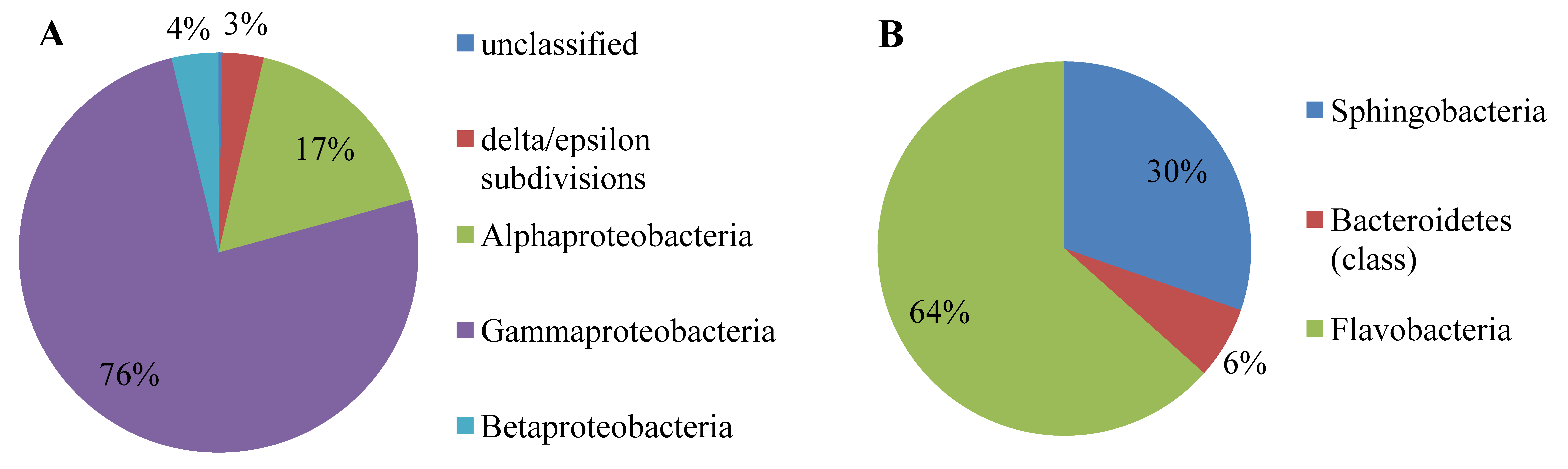
2.1. Metagenome Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Alignment Length | E-value | % Identity | Bit Score | Fragment (Start - End) | Taxonomy Assignment | Best Hit ID |
|---|---|---|---|---|---|---|---|
| Proteobacteria | |||||||
| contig16635 | 85 | 1.00E-40 | 100 | 168 | 1 - 85 | Sulfitobacter | 165917 |
| contig26360 | 150 | 2.51E-51 | 93 | 202 | 1 - 149 | Unclassified Rhodobacteraceae | 142124 |
| contig26572 | 162 | 7.94E-70 | 96 | 264 | 4 - 163 | Unclassified Rhodobacteraceae | 70710 |
| contig26707 | 130 | 2.51E-63 | 99 | 242 | 1 - 130 | Unclassified Rhodobacteraceae | 113926 |
| contig25574 | 235 | 2.51E-130 | 100 | 466 | 50 - 284 | Glaciecola | 108683 |
| contig26573 | 526 | 0.00E+00 | 99 | 1003 | 1 - 526 | Glaciecola | 80428 |
| contig26860 | 151 | 1.26E-80 | 100 | 299 | 1 - 151 | Glaciecola | 170839 |
| contig26820 | 696 | 0.00E+00 | 93 | 1015 | 6 - 701 | Teredinibacter | 144812 |
| contig00070 | 247 | 1.26E-81 | 93 | 305 | 738 - 983 | Cellvibrio | 98921 |
| contig12909 | 577 | 0.00E-00 | 93 | 858 | 209 - 784 | Unclassified Gammaproteobacteria | 151615 |
| Bacteriodetes | |||||||
| contig00061 | 892 | 0.00E+00 | 90 | 1094 | 1988 - 2879 | Roseivirga | 102384 |
| contig26228 | 139 | 2.51E-66 | 98 | 252 | 1 - 138 | Flavobacterium | 154970 |
| contig26430 | 241 | 5.01E-130 | 99 | 464 | 1 - 241 | Ulvibacter | 80102 |
| contig26765 | 436 | 5.01E-140 | 89 | 500 | 60 - 495 | Unclassified Cytophagaceae | 2577 |


2.2. Polysaccharide hydrolases
| CAZy family | Pfam ID | Number of hits |
|---|---|---|
| GH5 | PF00150 | 56 |
| GH6 | PF01341 | 5 |
| GH7 | PF00840 | 0 |
| GH8 | PF01270 | 40 |
| GH9 | PF00759 | 30 |
| GH12 | PF01670 | 1 |
| GH16 | PF00722 | 64 |
| GH18 | PF00704 | 10 |
| GH19 | PF00182 | 3 |
| GH45 | PF02015 | 0 |
| GH48 | PF02011 | 2 |
| GH61 | PF03443 | 0 |
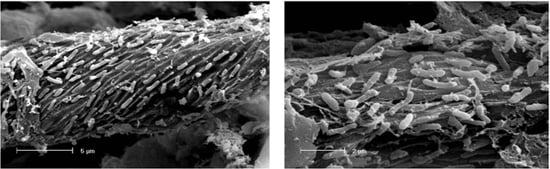
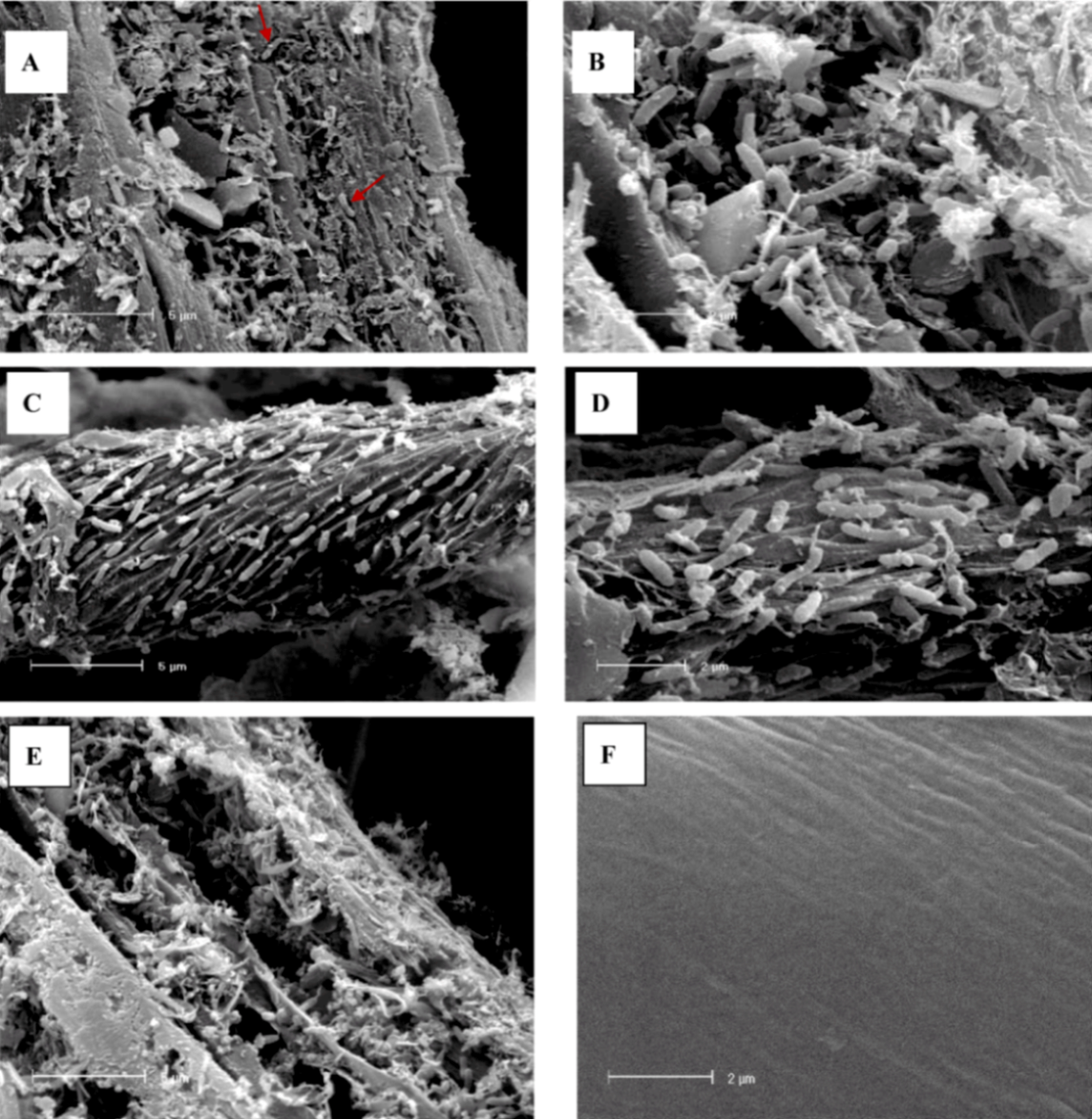
2.3. Scanning Electron Microscopy analysis of colonized cellulose bait
3. Experimental Section
3.1. Sampling

3.2. Metagenome Analysis
3.2.1. DNA extraction [42]
3.2.2. Pyrosequencing, sequence analysis
3.2.3. Glycoside Hydrolase database construction
3.3. Scanning Electron Microscopy (SEM) of colonized cellulose samples
4. Conclusions
Acknowledgements
Supplementary Files
References and Notes
- Nagata, T. Organic matter-bacteria interactions in seawater. In Microbial Ecology of the Oceans , 2nd; Kirchman, D.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- DeLong, E.F.; Karl, D.M. Genomic perspectives in microbial oceanography. Nature 2005, 437, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Delong, E.F.; Franks, D.G.; Alldredge, A.L. Phylogenetic diversity of aggregate-attached vs free-living marine bacterial assemblages. Limnol. Ocean. 1993, 38, 924–934. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; Delong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Garsoux, G.; Lamotte, J.; Gerday, C.; Feller, G. Kinetic, structural optimization to catalysis at low temperatures in a psychrophilic cellulase from the Antarctic bacterium Pseudoalteromonas haloplanktis. Biochem. J. 2004, 384, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Xiong, P.; Wen, J. Characterization, gene cloning of a cold-active cellulase from a deep-sea psychrotrophic bacterium Pseudoalteromonas sp. DY3. Extremophiles 2006, 10, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Weiner, R.M.; Taylor, L.E.; Henrissat, B.; Hauser, L.; Land, M.; Coutinho, P.M.; Rancurel, C.; Saunders, E.H.; Longmire, A.G.; Zhang, H.; et al. Complete genome sequence of the complex carbohydrate-degrading marine bacterium, Saccharophagus degradans strain 2-40 T . PLoS Genet. 2008, 4, e1000087. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Madupu, R.; Durkin, A.S.; Ekborg, N.A.; Pedamallu, C.S.; Hostetler, J.B.; Radune, D.; Toms, B.S.; Henrissat, B.; Coutinho, P.M.; et al. The complete genome of Teredinibacter turnerae T7901: an intracellular endosymbiont of marine wood-boring bivalves (shipworms) . PLoS One 2009, 4, e6085. [Google Scholar] [CrossRef] [PubMed]
- CAZy Home Page. Available online: http://www.cazy.org (accessed on 11 July 2010). [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics . Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Davies, G. Structural, sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Zhang, P.; Duan, C.J.; Mo, X.C.; Tang, J.L.; Feng, J.X. Identification of cellulase genes from the metagenomes of compost soils, functional characterization of one novel endoglucanase. Curr. Microbiol. 2009, 58, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, C.M.; Han, B.R.; Kim, M.Y.; Yeo, Y.S.; Yoon, S.H.; Koo, B.S.; Jun, H.K. Characterization of a gene encoding cellulase from uncultured soil bacteria. FEMS Microbiol. Lett. 2008, 282, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Allgaier, M.; Reddy, A.; Park, J.I.; Ivanova, N.; D'Haeseleer, P.; Lowry, S.; Sapra, R.; Hazen, T.C.; Simmons, B.A.; VanderGheynst, J.S.; et al. Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community . PLoS One 2010, 5, e8812. [Google Scholar] [CrossRef] [PubMed]
- Brulc, J.M.; Antonopoulos, D.A.; Miller, M.E.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, F.; Luginbuhl, P.; Ivanova, N.; Ghassemian, M.; Richardson, T.H.; Stege, J.T.; Cayouette, M.; McHardy, A.C.; Djordjevic, G.; Aboushadi, N.; et al. Metagenomic, functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450, 560–565. [Google Scholar] [CrossRef] [PubMed]
- de Menezes, A.B.; Lockhart, R.J.; Cox, M.J.; Allison, H.E.; McCarthy, A.J. Cellulose degradation by micromonosporas recovered from freshwater lakes, classification of these actinomycetes by DNA gyrase B gene sequencing. Appl. Environ. Microbiol. 2008, 74, 7080–7084. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.E.; de Menezes, A.B.; Allison, H.E.; McCarthy, A.J. Molecular biological detection, quantification of novel Fibrobacter populations in freshwater lakes. Appl. Environ. Microbiol. 2009, 75, 5148–5152. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D'Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server - a public resource for the automatic phylogenetic, functional analysis of metagenomes. BMC Bioinformatics 2008, 9, 386–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database, workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, E.F. Modern microbial seascapes. Nat. Rev. Microbiol. 2007, 5, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Rath, J.; Wu, K.Y.; Herndl, G.J.; DeLong, E.F. High phylogenetic diversity in a marine-snow-associated bacterial assemblage. Aquat. Microb. Ecol. 1998, 14, 261–269. [Google Scholar] [CrossRef]
- Simon, M.; Grossart, H.P.; Schweitzer, B.; Ploug, H. Microbial ecology of organic aggregates in aquatic ecosystems. Aquat. Microb. Ecol. 2002, 28, 175–211. [Google Scholar] [CrossRef]
- Smith, D.C.; Simon, M.; Alldredge, A.L.; Azam, F. Intense hydrolytic enzyme-activity on marine aggregates, implications for rapid particle dissolution. Nature 1992, 359, 139–142. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Stingl, U. Molecular diversity, ecology of microbial plankton. Nature 2005, 437, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crecy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation, its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Ekborg, N.A.; Gonzalez, J.M.; Howard, M.B.; Taylor, L.E.; Hutcheson, S.W.; Weiner, R.M. Saccharophagus degradans gen nov., sp nov., a versatile marine degrader of complex polysaccharides. Int. J. System. Evol. Microbiol. 2005, 55, 1545–1549. [Google Scholar] [CrossRef]
- Johansen, J.E.; Nielsen, P.; Sjoholm, C. Description of Cellulophaga baltica gen. nov., sp. nov., Cellulophaga fucicola gen. nov., sp. nov., reclassification of [Cytophaga] lytica to Cellulophaga lytica gen. nov., comb. nov. Int. J. Syst. Bacteriol. 1999, 49 (Pt 3), 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Kirchman, D.L. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [PubMed]
- Gomez-Pereira, P.R.; Fuchs, B.M.; Alonso, C.; Oliver, M.J.; van Beusekom, J.E.; Amann, R. Distinct flavobacterial communities in contrasting water masses of the north Atlantic Ocean. ISME J. 2010, 4, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, I.; Magnuson, J.K.; Collart, F.; Robbertse, B.; Adney, W.S.; Himmel, M.E.; Baker, S.E. Fungal glycoside hydrolases for saccharification of lignocellulose: outlook for new discoveries fueled by genomics, functional studies. Cellulose 2009, 16, 687–697. [Google Scholar] [CrossRef]
- Strohmeier, M.; Hrmova, M.; Fischer, M.; Harvey, A.J.; Fincher, G.B.; Pleiss, J. Molecular modeling of family GH16 glycoside hydrolases: potential roles for xyloglucan transglucosylases/hydrolases in cell wall modification in the poaceae. Protein Sci. 2004, 13, 3200–3213. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [PubMed]
- Xie, G.; Bruce, D.C.; Challacombe, J.F.; Chertkov, O.; Detter, J.C.; Gilna, P.; Han, C.S.; Lucas, S.; Misra, M.; Myers, G.L.; et al. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 2007, 73, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Pfam Home Page . Available online: http://pfam.sanger.ac.uk (accessed on 11 July 2010).
- Grimes, D.J.; Johnson, C.N.; Dillon, K.S.; Flowers, A.R.; Noriea, N.F.; Berutti, T. What genomic sequence information has revealed about Vibrio ecology in the ocean-v review. Microb. Ecol. 2009, 58, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.T.; Moore, J.A.; Kirchman, D.L. Chitinases from uncultured marine microorganisms. Appl. Environ. Microbiol. 1999, 65, 2553–2557. [Google Scholar] [PubMed]
- Elifantz, H.; Malmstrom, R.R.; Cottrell, M.T.; Kirchman, D.L. Assimilation of polysaccharides, glucose by major bacterial groups in the Delaware Estuary. Appl. Environ. Microbiol. 2005, 71, 7799–7805. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.T.; Yu, L.; Kirchman, D.L. Sequence, expression analyses of Cytophaga-like hydrolases in a western arctic metagenomic library, the Sargasso Sea. Appl. Environ. Microbiol. 2005, 71, 8506–8513. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Shimon, L.J.; Shoham, Y.; Lamed, R. Cellulosomes-structure, ultrastructure. J. Struct. Biol. 1998, 124, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R.I.; Whiteley, A.S.; O'Donnell, A.G.; Bailey, M.J. Rapid method for coextraction of DNA, RNA from natural environments for analysis of ribosomal DNA-, rRNA-based microbial community composition. Appl. Environ. Microbiol. 2000, 66, 5488–5491. [Google Scholar] [CrossRef] [PubMed]
- The SEED Home Page. Available online: http://metagenomics.theseed.org (accessed on 11 July 2010).
- Veltkamp, C.J.; Chubb, J.C.; Birch, S.P.; Eaton, J.W. A simple freeze dehydration method for studying epiphytic, epizoic communities using the scanning electron-microscope. Hydrobiologia 1994, 288, 33–38. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Hagstrom, A. Bacterial, archaeal community structure, its patterns. In Microbial Ecology of the OceansKirchman, D.L., Ed.; 2008, 2nd; John Wiley & Sons, Inc.: Hoboken, NJ, USA. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Edwards, J.L.; Smith, D.L.; Connolly, J.; McDonald, J.E.; Cox, M.J.; Joint, I.; Edwards, C.; McCarthy, A.J. Identification of Carbohydrate Metabolism Genes in the Metagenome of a Marine Biofilm Community Shown to Be Dominated by Gammaproteobacteria and Bacteroidetes. Genes 2010, 1, 371-384. https://doi.org/10.3390/genes1030371
Edwards JL, Smith DL, Connolly J, McDonald JE, Cox MJ, Joint I, Edwards C, McCarthy AJ. Identification of Carbohydrate Metabolism Genes in the Metagenome of a Marine Biofilm Community Shown to Be Dominated by Gammaproteobacteria and Bacteroidetes. Genes. 2010; 1(3):371-384. https://doi.org/10.3390/genes1030371
Chicago/Turabian StyleEdwards, Jennifer L., Darren L. Smith, John Connolly, James E. McDonald, Michael J. Cox, Ian Joint, Clive Edwards, and Alan J. McCarthy. 2010. "Identification of Carbohydrate Metabolism Genes in the Metagenome of a Marine Biofilm Community Shown to Be Dominated by Gammaproteobacteria and Bacteroidetes" Genes 1, no. 3: 371-384. https://doi.org/10.3390/genes1030371
APA StyleEdwards, J. L., Smith, D. L., Connolly, J., McDonald, J. E., Cox, M. J., Joint, I., Edwards, C., & McCarthy, A. J. (2010). Identification of Carbohydrate Metabolism Genes in the Metagenome of a Marine Biofilm Community Shown to Be Dominated by Gammaproteobacteria and Bacteroidetes. Genes, 1(3), 371-384. https://doi.org/10.3390/genes1030371