Environmental Conditions May Shape the Patterns of Genomic Variations in Leishmania panamensis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Clinical Sample Collection
2.3. Parasite Culture and Generation of an Antimony-Resistant L. panamensis Strain
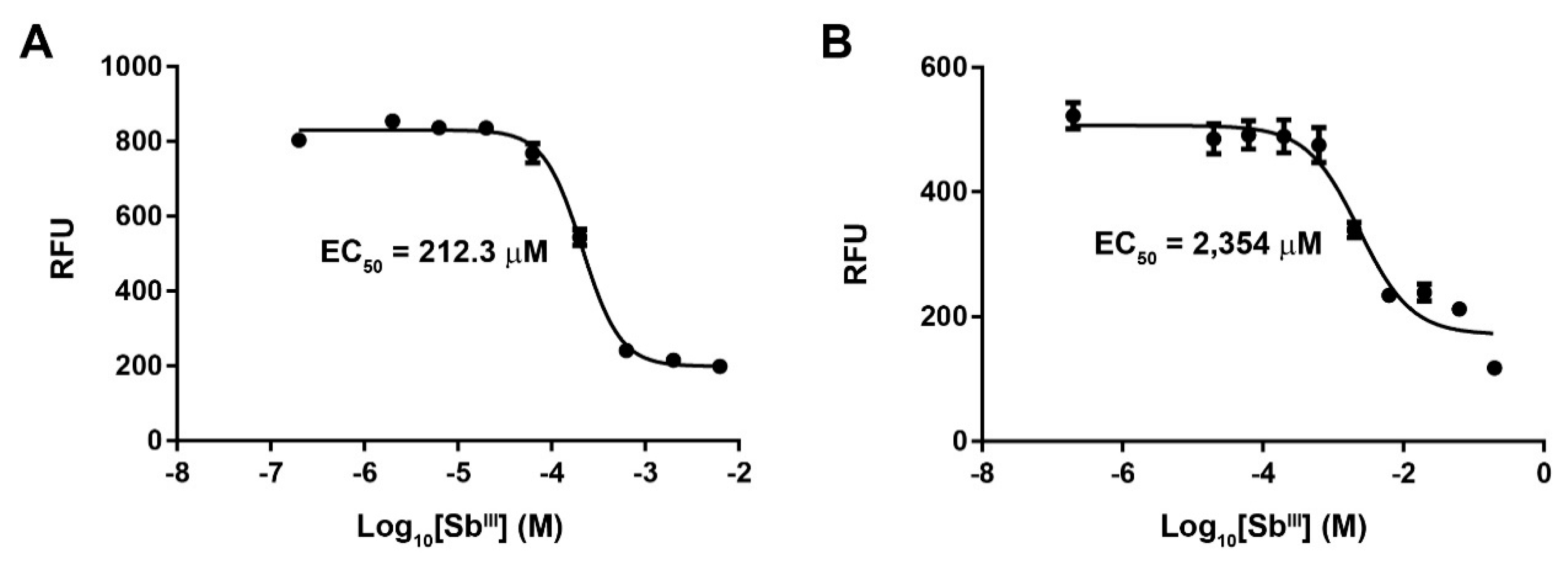
2.4. Antimony Susceptibility Assay
2.5. Whole Genome Sequencing
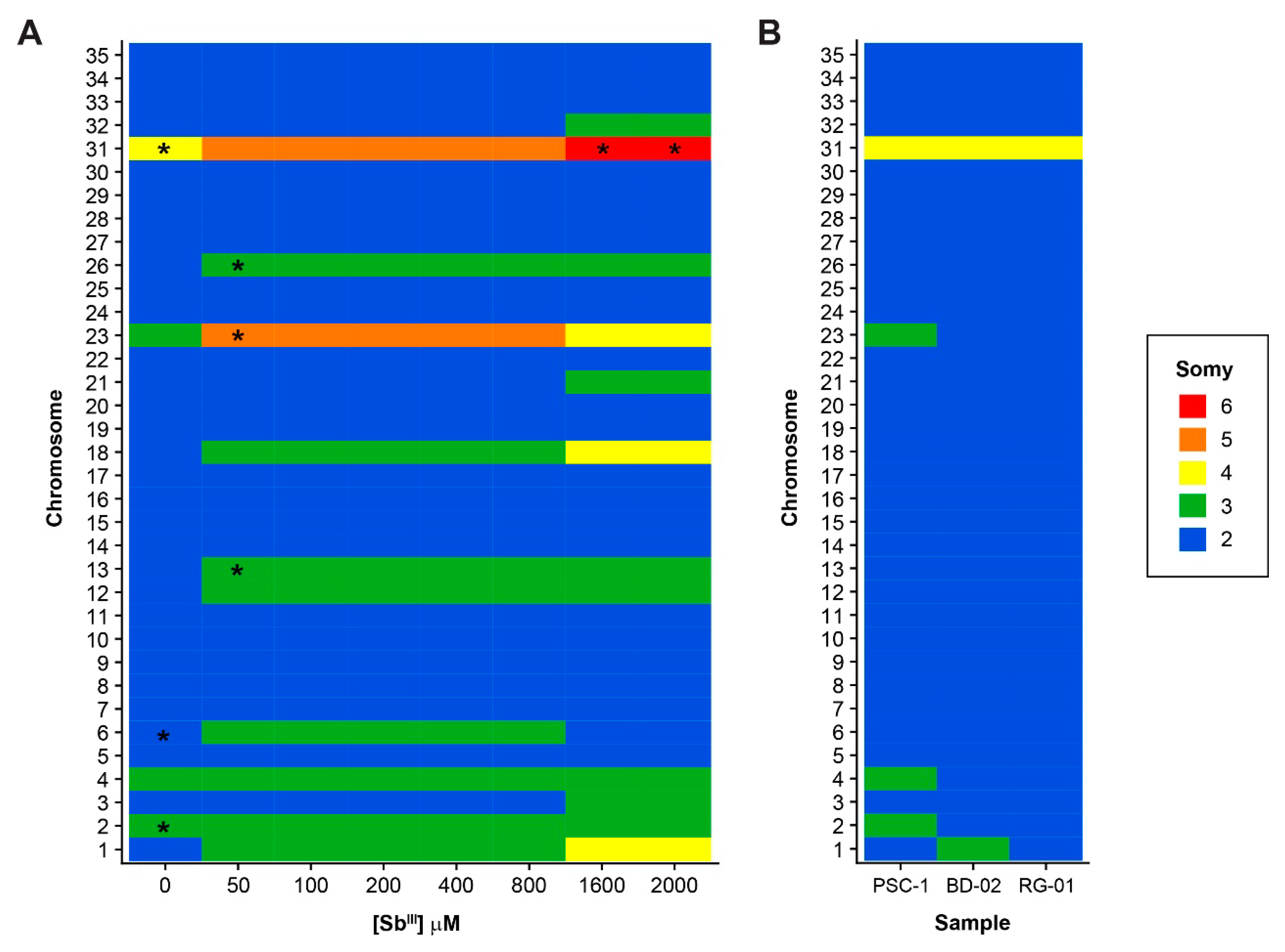
2.6. Estimation of Chromosome Somy
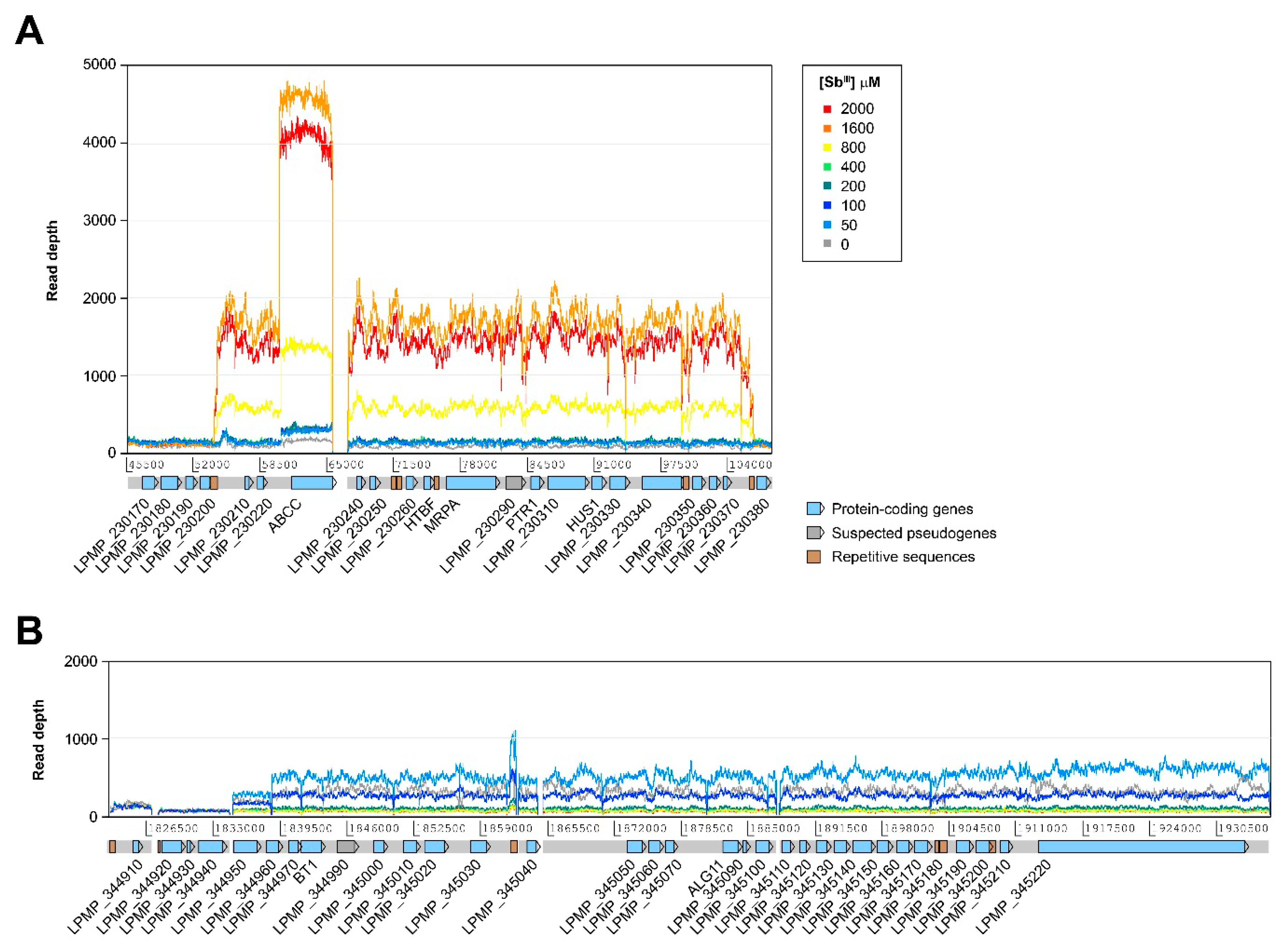
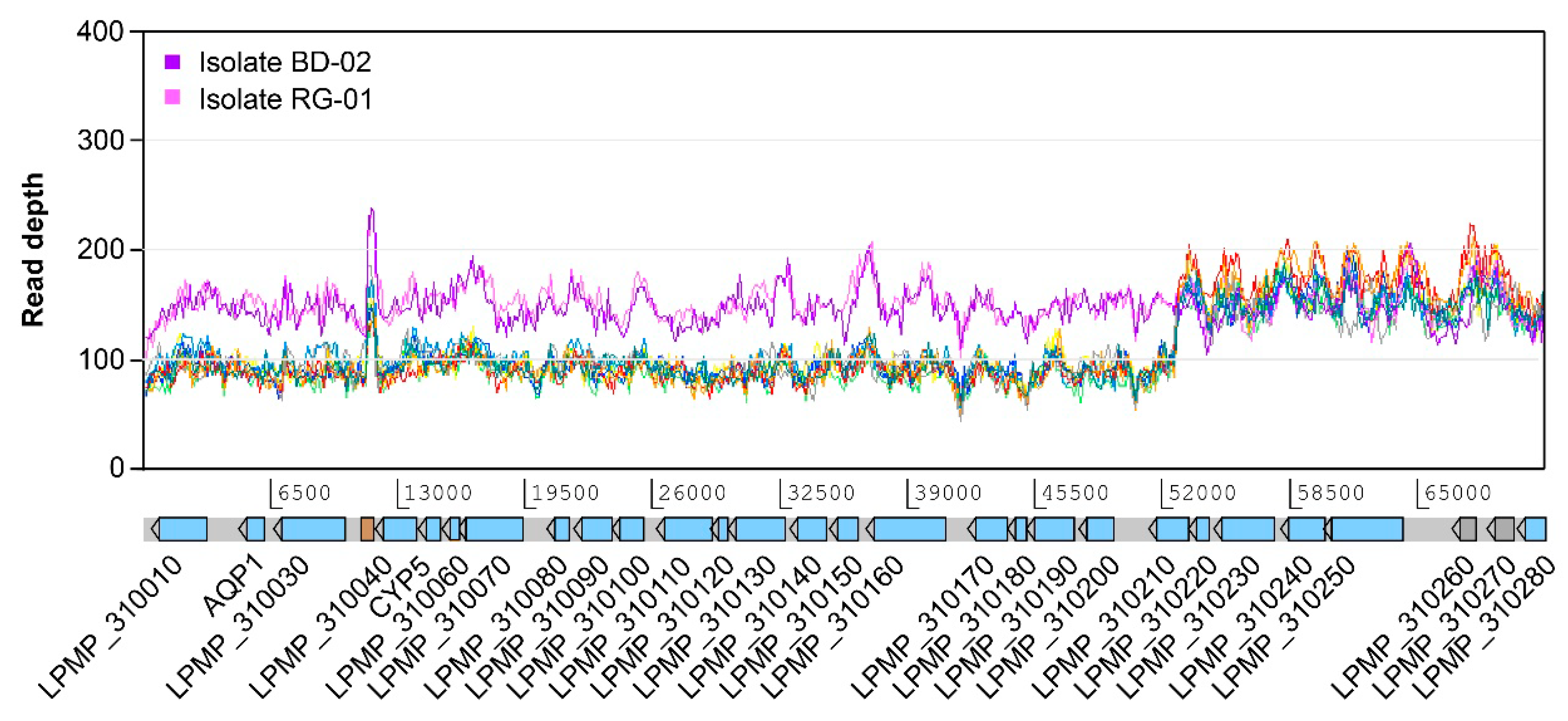
2.7. Detection of Genetic Amplifications
2.8. Detection of Variants Associated with Protein-Coding Genes
3. Results
3.1. Generation of an Antimony-Resistant L. panamensis Strain
3.2. Variations in Chromosome Somy and Relatively Large Chromosomal Regions
3.3. Variations at the Level of Individual Protein-Coding Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clayton, C. Gene expression in Kinetoplastids. Curr. Opin. Microbiol. 2016, 32, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Iantorno, S.A.; Durrant, C.; Khan, A.; Sanders, M.J.; Beverley, S.M.; Warren, W.C.; Berriman, M.; Sacks, D.L.; Cotton, J.A.; Grigg, M.E. Gene Expression in Leishmania Is Regulated Predominantly by Gene Dosage. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, J.-M.; Raymond, F.; Mukherjee, A.; Plourde, M.; Gingras, H.; Roy, G.; Lapointe, A.; Leprohon, P.; Papadopoulou, B.; Corbeil, J.; et al. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 2014, 12, e1001868. [Google Scholar] [CrossRef] [PubMed]
- Sterkers, Y.; Lachaud, L.; Bourgeois, N.; Crobu, L.; Bastien, P.; Pages, M.; Pagès, M. Novel insights into genome plasticity in Eukaryotes: Mosaic aneuploidy in Leishmania. Mol. Microbiol. 2012, 86, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Mannaert, A.; Vanaerschot, M.; Van der Auwera, G.; Dujardin, J.-C. (Post-) Genomic approaches to tackle drug resistance in Leishmania. Parasitology 2013, 140, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Leprohon, P.; Légaré, D.; Raymond, F.; Madore, E.; Hardiman, G.; Corbeil, J.; Ouellette, M. Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant Leishmania infantum. Nucleic Acids Res. 2009, 37, 1387–1399. [Google Scholar] [CrossRef]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, I.; Mottram, J.C.; et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef]
- Dumetz, F.; Imamura, H.; Sanders, M.; Seblova, V.; Myskova, J.; Pescher, P.; Vanaerschot, M.; Meehan, C.J.; Cuypers, B.; De Muylder, G.; et al. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio 2017. [Google Scholar] [CrossRef] [PubMed]
- Bussotti, G.; Gouzelou, E.; Boité, M.C.; Kherachi, I.; Harrat, Z.; Eddaikra, N.; Mottram, J.C.; Antoniou, M.; Christodoulou, V.; Bali, A.; et al. Leishmania genome dynamics during environmental adaptation reveal strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification. MBio 2018. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Boisvert, S.; Monte-Neto, R.L.D.; Coelho, A.C.; Raymond, F.; Mukhopadhyay, R.; Corbeil, J.; Ouellette, M. Telomeric gene deletion and intrachromosomal amplification in antimony-resistant Leishmania. Mol. Microbiol. 2013, 88, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Monte-Neto, R.; Laffitte, M.-C.N.; Leprohon, P.; Reis, P.; Frézard, F.; Ouellette, M. Intrachromosomal Amplification, Locus Deletion and Point Mutation in the Aquaglyceroporin AQP1 Gene in Antimony Resistant Leishmania (Viannia) guyanensis. PLoS Negl. Trop. Dis. 2015, 9, e0003476. [Google Scholar] [CrossRef]
- Patino, L.H.; Imamura, H.; Cruz-Saavedra, L.; Pavia, P.; Muskus, C.; Méndez, C.; Dujardin, J.C.; Ramírez, J.D. Major changes in chromosomal somy, gene expression and gene dosage driven by SbIII in Leishmania braziliensis and Leishmania panamensis. Sci. Rep. 2019, 9, 9485. [Google Scholar] [CrossRef]
- Dias, F.C.; Ruiz, J.C.; Lopes, W.C.Z.; Squina, F.M.; Renzi, A.; Cruz, A.K.; Tosi, L.R.O. Organization of H locus conserved repeats in Leishmania (Viannia) braziliensis correlates with lack of gene amplification and drug resistance. Parasitol. Res. 2007, 101, 667–676. [Google Scholar] [CrossRef]
- Haimeur, A.; Brochu, C.; Genest, P.A.; Papadopoulou, B.; Ouellette, M. Amplification of the ABC transporter gene PGPA and increased trypanothione levels in potassium antimonyl tartrate (SbIII) resistant Leishmania tarentolae. Mol. Biochem. Parasitol. 2000, 108, 131–135. [Google Scholar] [CrossRef]
- El Fadili, K.; Messier, N.; Leprohon, P.; Roy, G.; Guimond, C.; Trudel, N.; Saravia, N.G.; Papadopoulou, B.; Légaré, D.; Ouellette, M. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob. Agents Chemother. 2005. [Google Scholar] [CrossRef]
- Gourbal, B.; Sonuc, N.; Bhattacharjee, H.; Legare, D.; Sundar, S.; Ouellette, M.; Rosen, B.P.; Mukhopadhyay, R. Drug uptake and modulation of drug resistance in Leishmania by an aquaglyceroporin. J. Biol. Chem. 2004, 279, 31010–31017. [Google Scholar] [CrossRef]
- Segovia, M.; Ortiz, G. LD1 amplifications in Leishmania. Parasitol. Today 1997, 13, 196–199. [Google Scholar] [CrossRef]
- Sunkin, S.M.; McDonagh, P.; Cunningham, M.L.; Beverley, S.M.; Stuart, K.; Myler, P.J. Conservation of the LD1 region in Leishmania includes DNA implicated in LD1 amplification. Mol. Biochem. Parasitol. 2001, 113, 315–321. [Google Scholar] [CrossRef]
- Fu, G.; Melville, S.; Brewster, S.; Warner, J.; Barker, D.C. Analysis of the genomic organisation of a small chromosome of Leishmania braziliensis M2903 reveals two genes encoding GTP-binding proteins, one of which belongs to a new G-protein family and is an antigen. Gene 1998, 210, 325–333. [Google Scholar] [CrossRef]
- Llanes, A.; Restrepo, C.M.; Del Vecchio, G.; Anguizola, F.J.; Lleonart, R.; Del Vecchio, G.; Anguizola, F.J.; Lleonart, R. The genome of Leishmania panamensis: Insights into genomics of the L. (Viannia) subgenus. Sci. Rep. 2015, 5, 8550. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, H.; Downing, T.; van den Broeck, F.; Sanders, M.J.; Rijal, S.; Sundar, S.; Mannaert, A.; Vanaerschot, M.; Berg, M.; de Muylder, G.; et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife 2016. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wala, J.A.; Bandopadhayay, P.; Greenwald, N.F.; O’Rourke, R.; Sharpe, T.; Stewart, C.; Schumacher, S.; Li, Y.; Weischenfeldt, J.; Yao, X.; et al. SvABA: Genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018. [Google Scholar] [CrossRef]
- Mohapatra, S. Drug resistance in leishmaniasis: Newer developments. Trop. Parasitol. 2014, 4, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.C.; Fase-Fowler, F.; van Luenen, H.; Calafat, J.; Borst, P. The H circles of Leishmania tarentolae are a unique amplifiable system of oligomeric DNAs associated with drug resistance. J. Biol. Chem. 1988, 263, 16977–16983. [Google Scholar]
- Ouellette, M.; Fase-Fowler, F.; Borst, P. The amplified H circle of methotrexate-resistant Leishmania tarentolae contains a novel P-glycoprotein gene. EMBO J. 1990, 9, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Gamarro, F.; Chiquero, M.J.; Amador, M.V.; Légaré, D.; Ouellette, M.; Castanys, S. P-glycoprotein overexpression in methotrexate-resistant Leishmania tropica. Biochem. Pharmacol. 1994, 47, 1939–1947. [Google Scholar] [CrossRef]
- Downing, T.; Stark, O.; Vanaerschot, M.; Imamura, H.; Sanders, M.; Decuypere, S.; de Doncker, S.; Maes, I.; Rijal, S.; Sundar, S.; et al. Genome-wide SNP and microsatellite variation illuminate population-level epidemiology in the Leishmania donovani species complex. Infect Genet. Evol. 2012, 12, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Légaré, D.; Richard, D.; Mukhopadhyay, R.; Stierhof, Y.D.; Rosen, B.P.; Haimeur, A.; Papadopoulou, B.; Ouellette, M. The Leishmania ATP-binding Cassette Protein PGPA is an Intracellular Metal-Thiol Transporter ATPase. J. Biol. Chem. 2001, 276, 26301–26307. [Google Scholar] [CrossRef] [PubMed]
- Frézard, F.; Monte-Neto, R.; Reis, P.G. Antimony transport mechanisms in resistant Leishmania parasites. Biophys. Rev. 2014, 6, 119–132. [Google Scholar] [CrossRef]
- Leprohon, P.; Fernandez-Prada, C.; Gazanion, É.; Monte-Neto, R.; Ouellette, M. Drug resistance analysis by next generation sequencing in Leishmania. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 26–35. [Google Scholar] [CrossRef]
- Barja, P.P.; Pescher, P.; Bussotti, G.; Dumetz, F.; Imamura, H.; Kedra, D.; Domagalska, M.; Chaumeau, V.; Himmelbauer, H.; Pages, M.; et al. Haplotype selection as an adaptive mechanism in the protozoan pathogen Leishmania donovani. Nat. Ecol. Evol. 2017. [Google Scholar] [CrossRef]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; Amon, A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Torres, E.M.; Dunham, M.J.; Amon, A. Transcriptional consequences of aneuploidy. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [PubMed]
- Brauer, M.J.; Huttenhower, C.; Airoldi, E.M.; Rosenstein, R.; Matese, J.C.; Gresham, D.; Boer, V.M.; Troyanskaya, O.G.; Botstein, D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 2008. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000. [Google Scholar] [CrossRef] [PubMed]
- Regenberg, B.; Grotkjær, T.; Winther, O.; Fausbøll, A.; Åkesson, M.; Bro, C.; Hansen, L.K.; Brunak, S.; Nielsen, J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Amon, A. Gene copy-number alterations: A cost-benefit analysis. Cell 2013, 152, 394–405. [Google Scholar] [CrossRef]
- Katz, W.; Weinstein, B.; Solomon, F. Regulation of tubulin levels and microtubule assembly in Saccharomyces cerevisiae: Consequences of altered tubulin gene copy number. Mol. Cell. Biol. 1990. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Suspected Function | Estimated Haploid Copy Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PSC-1 | Treated Samples [SbIII] µM | RG-01 | BD-02 | ||||||||
| 50 | 100 | 200 | 400 | 800 | 1600 | 2000 | |||||
| LPMP_020120 | Phosphoglycan β-1,3-galactosyltransferase | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 5 |
| LPMP_040170 | Hypothetical protein | 10 | 14 | 14 | 14 | 14 | 14 | 13 | 13 | 18 | 14 |
| LPMP_040280 | β-fructofuranosidase | 3 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 2 | 5 |
| LPMP_050490 | ATPase α subunit | 3 | 4 | 4 | 4 | 4 | 4 | 3 | 3 | 4 | 6 |
| LPMP_060560 | 60S ribosomal protein L23a | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| LPMP_080610 | Amastin-like protein | 14 | 19 | 17 | 17 | 17 | 18 | 16 | 16 | 14 | 21 |
| LPMP_080680 | Tuzin | 22 | 30 | 27 | 27 | 26 | 28 | 27 | 26 | 39 | 34 |
| LPMP_080890 | Hypothetical protein | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| LPMP_090170 | Autophagy-related protein 8 (ATG8) | 13 | 19 | 20 | 19 | 20 | 21 | 19 | 19 | 20 | 16 |
| LPMP_100410 | GP63, leishmanolysin | 9 | 12 | 12 | 11 | 11 | 11 | 10 | 11 | 13 | 10 |
| LPMP_100440 | GP63, leishmanolysin | 6 | 8 | 8 | 8 | 8 | 8 | 7 | 7 | 8 | 7 |
| LPMP_130280 | α-tubulin | 19 | 21 | 22 | 22 | 23 | 22 | 22 | 22 | 26 | 24 |
| LPMP_160880 | Flagellar calcium-binding protein | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| LPMP_170080 | Elongation factor 1-α | 15 | 21 | 21 | 21 | 21 | 22 | 20 | 20 | 33 | 34 |
| LPMP_190790 | Autophagy-related protein 8 (ATG8) | 10 | 15 | 15 | 16 | 14 | 16 | 14 | 14 | 11 | 16 |
| LPMP_311770 | Ubiquitin-fusion protein | 32 | 28 | 27 | 27 | 27 | 28 | 22 | 23 | 51 | 50 |
| LPMP_330370 | Heat shock protein 83 | 12 | 14 | 14 | 14 | 14 | 14 | 13 | 13 | 16 | 12 |
| LPMP_330860 | β-tubulin | 34 | 45 | 43 | 43 | 43 | 44 | 41 | 41 | 41 | 34 |
| LPMP_331700 | Peptidase M20/M25/M40 | 12 | 15 | 14 | 15 | 15 | 14 | 14 | 14 | 15 | 12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Restrepo, C.M.; Llanes, A.; Cedeño, E.M.; Chang, J.H.; Álvarez, J.; Ríos, M.; Penagos, H.; Suárez, J.A.; Lleonart, R. Environmental Conditions May Shape the Patterns of Genomic Variations in Leishmania panamensis. Genes 2019, 10, 838. https://doi.org/10.3390/genes10110838
Restrepo CM, Llanes A, Cedeño EM, Chang JH, Álvarez J, Ríos M, Penagos H, Suárez JA, Lleonart R. Environmental Conditions May Shape the Patterns of Genomic Variations in Leishmania panamensis. Genes. 2019; 10(11):838. https://doi.org/10.3390/genes10110838
Chicago/Turabian StyleRestrepo, Carlos M., Alejandro Llanes, Eymi M. Cedeño, Jim H. Chang, Jennifer Álvarez, Margarita Ríos, Homero Penagos, José A. Suárez, and Ricardo Lleonart. 2019. "Environmental Conditions May Shape the Patterns of Genomic Variations in Leishmania panamensis" Genes 10, no. 11: 838. https://doi.org/10.3390/genes10110838
APA StyleRestrepo, C. M., Llanes, A., Cedeño, E. M., Chang, J. H., Álvarez, J., Ríos, M., Penagos, H., Suárez, J. A., & Lleonart, R. (2019). Environmental Conditions May Shape the Patterns of Genomic Variations in Leishmania panamensis. Genes, 10(11), 838. https://doi.org/10.3390/genes10110838