Search for Ancestral Features in Genomes of Rhizobium leguminosarum bv. viciae Strains Isolated from the Relict Legume Vavilovia formosa


 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
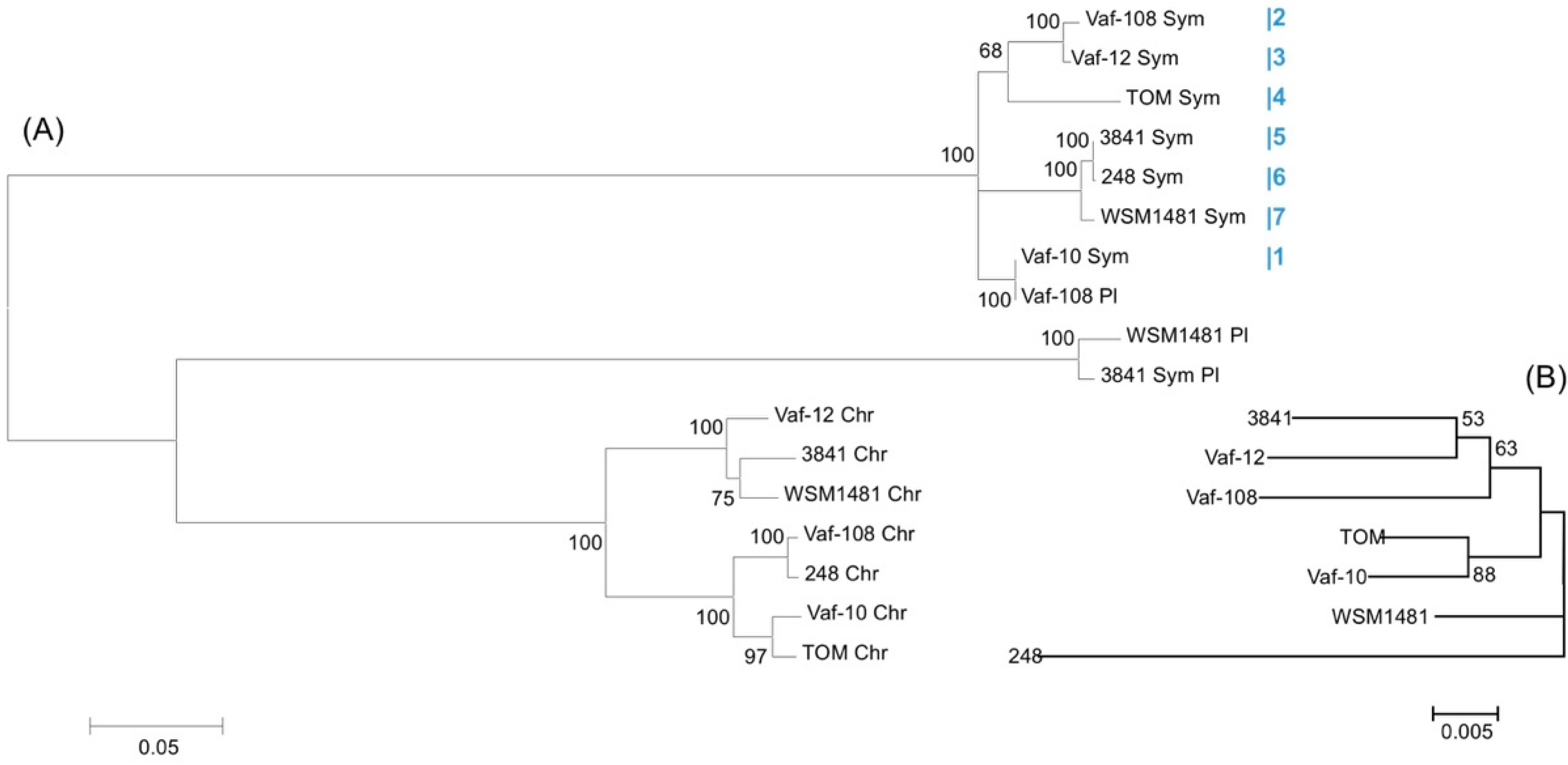
3.1. Vavilovia’s Isolates in Context of Core Genome of R. leguminosarum bv. viciae
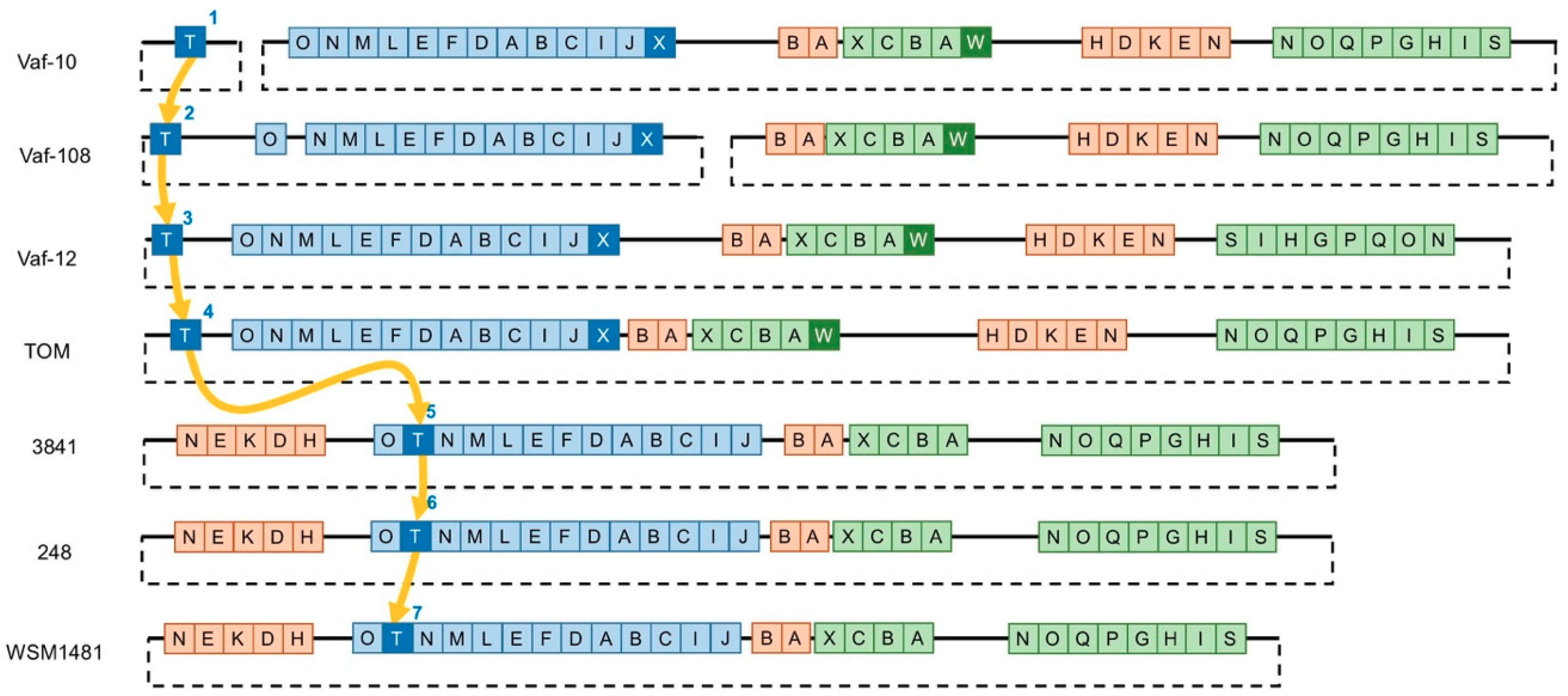
3.2. Variability of the Sym Gene Arrangement
3.3. Gain-and-Loss of Sym Genes
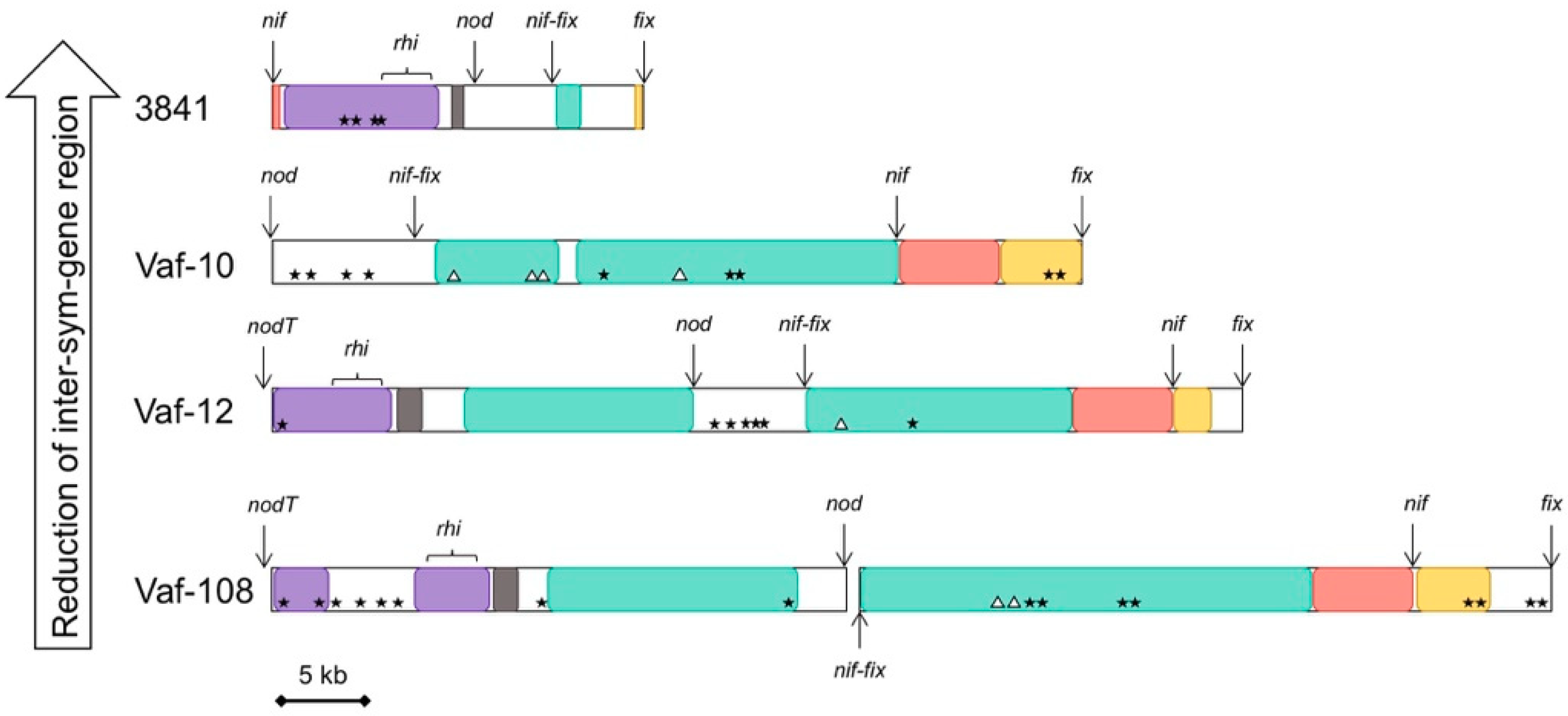
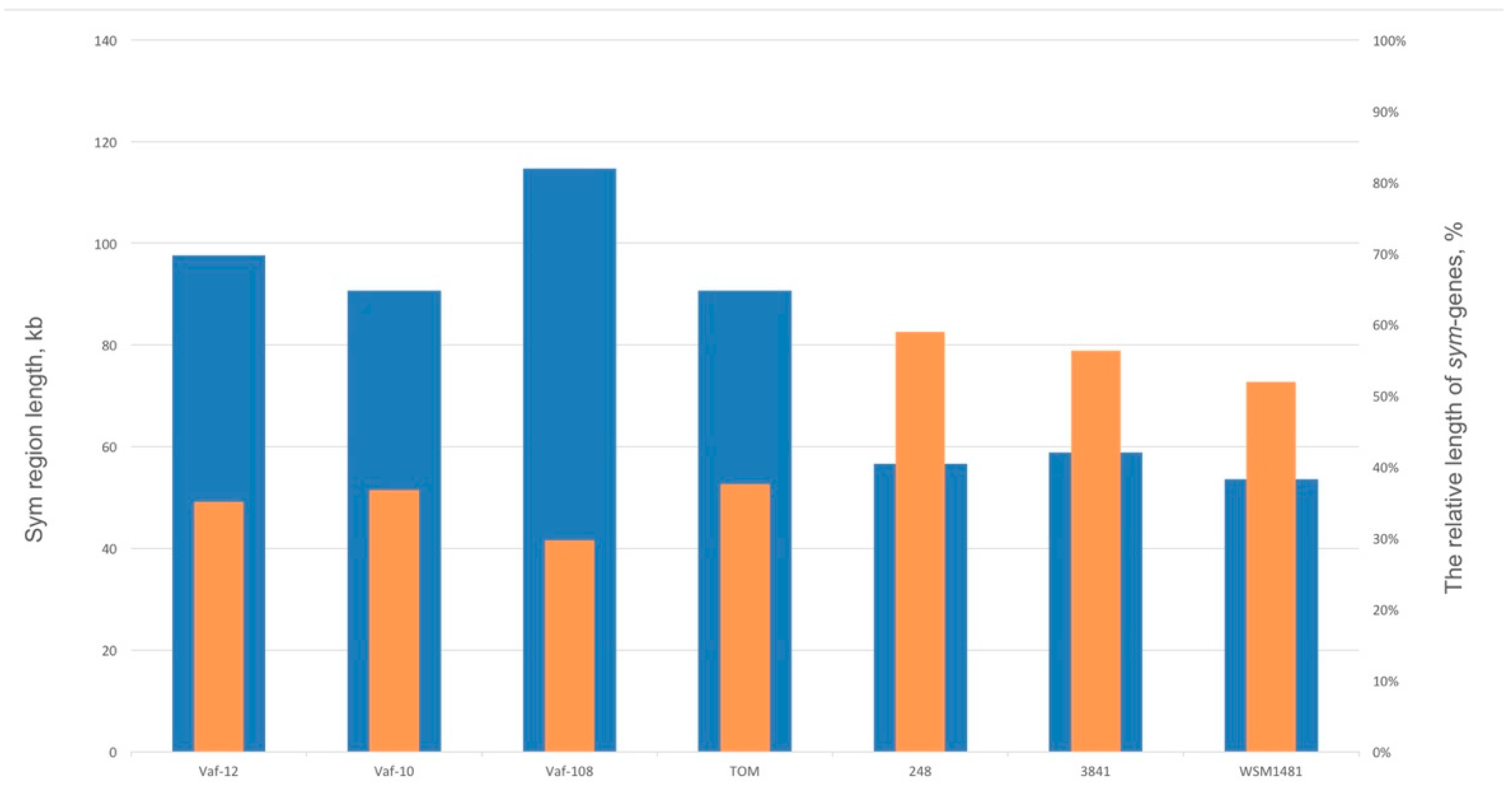
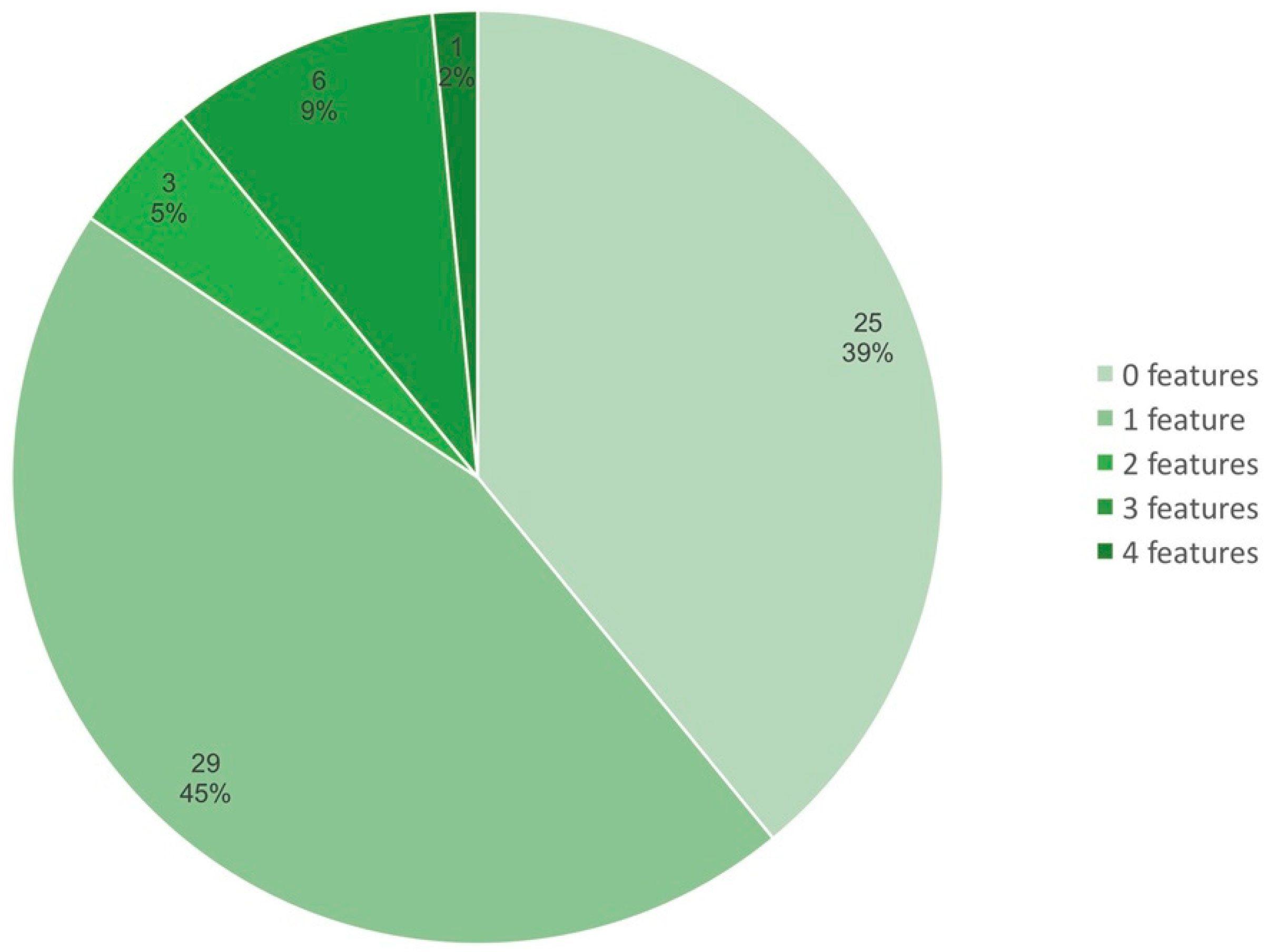
3.4. Compaction of the Sym Gene Cluster
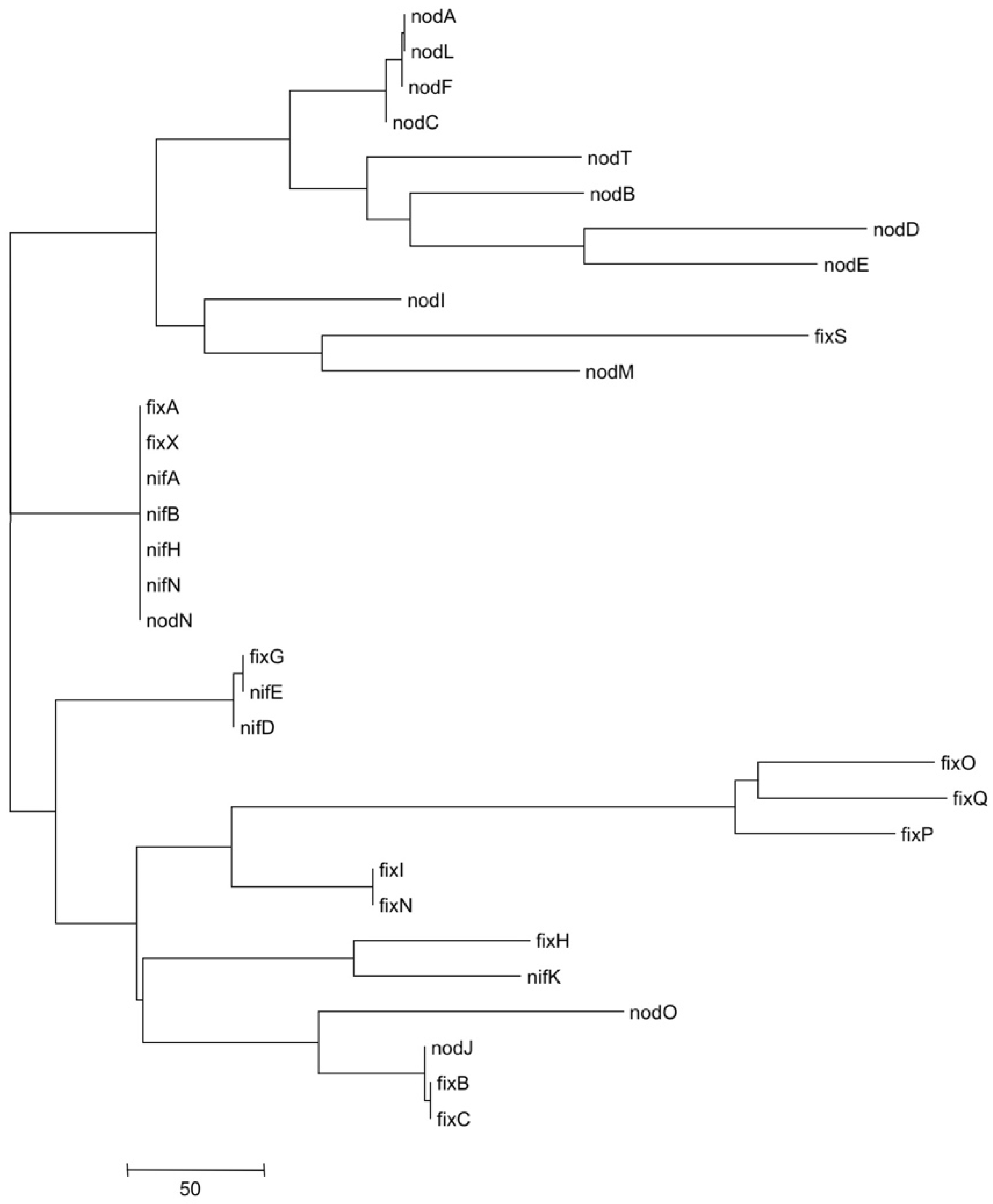
3.5. Metatree Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Young, J.P.W. Phylogeny and taxonomy of rhizobia. Plant Soil 1996, 186, 45–52. [Google Scholar] [CrossRef]
- Zakhia, F.; de Lajudie, P. Taxonomy of rhizobia. Agronomie 2001, 21, 569–576. [Google Scholar] [CrossRef]
- Gottfert, M. Regulation and function of rhizobial nodulation genes. FEMS Microbiol. Rev. 1993, 104, 39–64. [Google Scholar] [CrossRef] [PubMed]
- Debellé, F.; Moulin, L.; Mangin, B.; Dénarié, J.; Boivin, C. Nod genes and Nod signals and the evolution of the Rhizobium legume symbiosis. Acta Biochim. Pol. 2001, 48, 359–365. [Google Scholar] [PubMed]
- Shamseldin, A. The Role of Different Genes Involved in Symbiotic Nitrogen Fixation—Review. GJBB 2013, 8, 84–94. [Google Scholar] [CrossRef]
- Fischer, H.M. Genetic Regulation of Nitrogen Fixation in Rhizobia. Microbiol. Rev. 1994, 58, 352–386. [Google Scholar]
- Provorov, N.A.; Andronov, E.E. Evolution of root nodule bacteria: Reconstruction of the speciation processes resulting from genomic rearrangements in a symbiotic system. Microbiology 2016, 85, 131–139. [Google Scholar] [CrossRef]
- Rey, F.E.; Harwood, C.S. FixK, a global regulator of microaerobic growth, controls photosynthesis in Rhodopseudomonas palustris. Mol. Microbiol. 2010, 75, 1007–1020. [Google Scholar] [CrossRef]
- Wongdee, J.; Boonkerd, N.; Teaumroong, N.; Tittabutr, P.; Giraud, E. Regulation of Nitrogen Fixation in Bradyrhizobium sp. Strain DOA9 Involves Two Distinct NifA Regulatory Proteins That Are Functionally Redundant During Symbiosis but Not During Free-Living Growth. Front. Microbiol. 2018, 9, 1644. [Google Scholar] [CrossRef]
- Hassan, S.; Mathesius, U. The role of flavonoids in root-rhizosphere signalling: Opportunities and challenges for improving plant-microbe interactions. J. Exp. Bot. 2012, 63, 3429–3444. [Google Scholar] [CrossRef]
- Terpolilli, J.J.; Hood, G.A.; Poole, P.S. What determines the efficiency of N(2)-fixing Rhizobium-legume symbioses? Adv. Microb. Physiol. 2012, 60, 325–389. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Noisangiam, R.; Okubo, T.; Kaneko, T.; Oshima, K.; Hattori, M. Genome analysis of a novel Bradyrhizobium sp. DOA9 carrying a symbiotic plasmid. PLoS ONE 2015, 10, e0117392. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.T.; Trzebiatowski, J.R.; Cruickshank, R.W.; Gouzy, J.; Brown, S.D.; Elliot, R.M. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J. Bacteriol. 2002, 184, 3086–3095. [Google Scholar] [CrossRef] [PubMed]
- Poole, P.; Ramachandran, V.; Terpolilli, J. Rhizobia: From saprophytes to endosymbionts. Nat. Rev. Microbiol. 2018, 16, 291–303. [Google Scholar] [CrossRef]
- Andrews, M.; Andrews, M.E. Specificity in Legume-Rhizobia Symbioses. Int. J. Mol. Sci. 2017, 18, 705. [Google Scholar] [CrossRef]
- Safronova, V.I.; Kimeklis, A.K.; Chizhevskaya, E.P.; Belimov, A.A.; Andronov, E.E.; Pinaev, A.G.; Pukhaev, A.R.; Popov, K.P.; Tikhonovich, I.A. Genetic diversity of rhizobia isolated from nodules of the relic species Vavilovia formosa (Stev.) Fed. Antonie Leeuwenhoek 2014, 105, 389–399. [Google Scholar] [CrossRef]
- Mikić, A.; Smýkal, P.; Kenicer, G.; Vishnyakova, M.; Sarukhanyan, N.; Akopian, J.A.; Vanyan, A.; Gabrielyan, I.; Smýkalová, I.; Sherbakova, E.; et al. Beauty will save the world, but will the world save beauty? The case of the highly endangered Vavilovia formosa (Stev.) Fed. Planta 2014, 240, 1139–1146. [Google Scholar] [CrossRef]
- Govorov, L.I. Peas. In Flora of Cultivated Plants; Vavilov, N.I., Wulff, E.V., Eds.; Kolos: Leningrad, Russia, 1937; Volume 4, pp. 231–336. (In Russian) [Google Scholar]
- Kimeklis, A.K.; Chirak, E.R.; Kuznetsova, I.G.; Sazanova, A.L.; Safronova, V.I.; Belimov, A.A.; Onishchuk, O.P.; Kurchak, O.N.; Aksenova, T.S.; Pinaev, A.G.; et al. Rhizobia isolated from the relict legume Vavilovia formosa represent a genetically specific group within Rhizobium leguminosarum bv. viciae. Genes. under review.
- Kimeklis, A.K.; Safronova, V.I.; Kuznetsova, I.G.; Sazanova, A.L.; Belimov, A.A.; Pinaev, A.G.; Chizhevskaya, E.P.; Pukhaev, A.R.; Popov, K.P.; Andronov, E.E.; et al. Phylogenetic analysis of Rhizobium strains isolated from root nodules of Vavilovia formosa (Stev.) Fed. Sel’skokhozyaistvennaya Biol. [Agric. Biol.] 2015, 50, 655–664. [Google Scholar] [CrossRef]
- Davis, E.O.; Evans, I.J.; Johnston, A.W.B. Identification of nodX, a gene that allows Rhizobium leguminosarum biovar viciae strain TOM to nodulate Afghanistan peas. Mol. Gen. Genet. 1988, 212, 531–535. [Google Scholar] [CrossRef]
- Kimeklis, A.K.; Kuznetsova, I.G.; Sazanova, A.L.; Safronova, V.I.; Belimov, A.A.; Onishchuk, O.P.; Kurchak, O.N.; Aksenova, T.S.; Pinaev, A.G.; Musaev, A.M.; et al. Divergent Evolution of Symbiotic Bacteria: Rhizobia of the Relic Legume Vavilovia formosa Form an Isolated Group within Rhizobium leguminosarum bv. viciae. Russ. J. Genet. 2018, 54, 866–870. [Google Scholar] [CrossRef]
- Novikova, N.; Safronova, V. Transconjugants of Agrobacterium radiobacter harbouring sym genes of Rhizobium galegae can form an effective symbiosis with Medicago sativa. FEMS Microbiol. Lett. 1992, 93, 261–268. [Google Scholar] [CrossRef]
- Somasegaran, P.; Hoben, H.J. Isolating and Purifying Genomic DNA of Rhizobia Using a Large-Scale Method. In Handbook for Rhizobia: Methods in Legume-Rhizobium Technology; Springer: New York, NY, USA, 1994; pp. 279–283. [Google Scholar] [CrossRef]
- Safronova, V.; Tikhonovich, I. Automated cryobank of microorganisms: Unique possibilities for long-term authorized depositing of commercial microbial strains. In Microbes in Applied Research: Current Advances and Challenges; Mendez-Vilas, A., Ed.; World Scientific Publishing Co.: Singapore, 2012; pp. 331–334. [Google Scholar] [CrossRef]
- Russian Collection of Agricultural Microorganisms (RCAM). Available online: http://www.arriam.spb.ru/eng/lab10/ (accessed on 1 October 2019).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- HGAP: Hierarchical Genome Assembly Process. Available online: https://github.com/ben-lerch/HGAP-3.0 (accessed on 30 May 2018).
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A Customizable Web Server for Fast Metagenomic Sequence Analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef]
- Chirak, E.R.; Kopat’, V.V.; Kimeklis, A.K.; Safronova, V.I.; Belimov, A.A.; Chirak, E.L.; Tupikin, A.E.; Andronov, E.E.; Provorov, N.A. Structural and Functional Organization of the Plasmid Regulons of Rhizobium leguminosarum Symbiotic Genes. Microbiology 2016, 85, 708–716. [Google Scholar] [CrossRef]
- Reeve, W.; Ardley, J.; Tian, R.; Eshragi, L.; Yoon, J.W.; Ngamwisetkun, P.; Seshadri, R.; Ivanova, N.N.; Kyrpides, N.C. A Genomic Encyclopedia of the Root Nodule Bacteria: Assessing genetic diversity through a systematic biogeographic survey. Stand. Genomic. Sci. 2015, 10, 14. [Google Scholar] [CrossRef]
- Young, J.P.W.; Crossman, L.C.; Johnston, A.W.B.; Thomson, N.R.; Ghazoui, Z.F.; Hull, K.H.; Wexler, M.; Curson, A.R.; Todd, J.D.; Poole, P.S.; et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7, R34. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Nye, T.M.W.; Liò, P.; Gilks, W.R. A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics 2006, 22, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- GNU Parallel 2018. Available online: https://zenodo.org/record/1146014#.XdULkxMzbVp (accessed on 20 November 2019).
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Rodelas, B.; Lithgow, J.K.; Wisniewski-Dye, F.; Hardman, A.; Wilkinson, A.; Economou, A.; Williams, P.; Downie, J.A. Analysis of quorum-sensing-dependent control of rhizosphere-expressed (rhi) genes in Rhizobium leguminosarum bv. viciae. J. Bacteriol. 1999, 181, 3816–3823. [Google Scholar] [PubMed]
- Onishchuk, O.P.; Vorobyov, N.I.; Provorov, N.A. Nodulation competitiveness of nodule bacteria: Genetic control and adaptive significance: Review. Prikl. Biokhim. Mikrobiol. 2017, 53, 131–139. [Google Scholar] [CrossRef]
- Vavilov, N.I. Centers of origin of cultivated plants. Trends Pract. Bot. Genet. Sel. 1926, 16, 3–248. (In Russian) [Google Scholar]
- Zhukovskii, P.M. New origin and genetic centers of crops and highly endemic microcenters of related species. Bot. Z. 1968, 53, 430–460. (In Russian) [Google Scholar]
- Makasheva, R.H. Peas; Kolos: Leningrad, Russia, 1973; pp. 28–87. (In Russian) [Google Scholar]
- Sprent, J.I.; Ardley, J.; James, E.K. Biogeography of nodulated legumes and their nitrogen-fixing symbionts. New Phytol. 2017, 25, 40–56. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Ortega, C.; Olivares, J.; Martínez, J.L. RND multidrug efflux pumps: What are they good for? Front. Microbiol. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Mendoza, A.; Nava, N.; Santana, O.; Abreu-Goodger, C.; Tovar, A.; Quinto, C. Diminished Redundancy of Outer Membrane Factor Proteins in Rhizobiales: A nodT Homolog Is Essential for Free-Living Rhizobium etli. J. Mol. Microbiol. Biotechnol. 2007, 13, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Myllykallio, H.; Liebl, U. Dual role for cytochrome cbb3 oxidase in clinically relevant proteobacteria. Trends Microbiol. 2000, 8, 542–543. [Google Scholar] [CrossRef]
- Talbi, C.; Sanchez, C.; Hidalgo-Garcia, A.; González, E.M.; Arrese-Igor, C.; Girard, L.; Bedmar, E.J.; Delgado, M.J. Enhanced expression of Rhizobium etli cbb3 oxidase improves drought tolerance of common bean symbiotic nitrogen fixation. J. Exp. Bot. 2012, 63, 5035–5043. [Google Scholar] [CrossRef] [PubMed]
- Kopat, V.V.; Chirak, E.R.; Kimeklis, A.K.; Safronova, V.I.; Belimov, A.A.; Kabilov, M.R.; Andronov, E.E.; Provorov, N.A. Evolution of fixNOQP Genes Encoding Cytochrome Oxidase with High Affinity to Oxygen in Rhizobia and Related Bacteria. Russ. J. Genet. 2017, 53, 766–774. [Google Scholar] [CrossRef]
- Provorov, N.A.; Andronov, E.E.; Onishchuk, O.P. Forms of natural selection controlling the genomic evolution in nodule bacteria. Russ. J. Genet. 2017, 53, 411–419. [Google Scholar] [CrossRef]
- Abicht, H.K.; Schärer, M.A.; Quade, N.; Ledermann, R.; Mohorko, E.; Capitani, G.; Hennecke, H.; Glockshuber, R. How Periplasmic Thioredoxin TlpA Reduces Bacterial Copper Chaperone ScoI and Cytochrome Oxidase Subunit II (CoxB) Prior to Metallation. J. Biol. Chem. 2014, 289, 32431–32444. [Google Scholar] [CrossRef] [Green Version]
- Hontelez, J.G.; Lankhorst, R.K.; Katinakis, P.; van den Bos, R.C.; van Kammen, A. Characterization and nucleotide sequence of a novel gene fixW upstream of the fixABC operon in Rhizobium leguminosarum. Mol. Gen. Genet. 1989, 218, 536–544. [Google Scholar] [CrossRef]
- Wang, D.; Griffitts, J.; Starker, C.; Fedorova, E.; Limpens, E.; Ivanov, S.; Bisseling, T.; Long, S. A Nodule-Specific Protein Secretory Pathway Required for Nitrogen-Fixing Symbiosis. Science 2010, 327, 1126–1129. [Google Scholar] [CrossRef] [Green Version]
- Tsyganova, A.V.; Seliverstova, E.V.; Onischuk, O.P.; Kurchak, O.N.; Kimeklis, A.K.; Sazanova, A.L.; Kuznetsova, I.G.; Safronova, V.I.; Belimov, A.A.; Andronov, E.E.; et al. Diverse Ultrastructural Features of Symbiotic Nodules from Relict Legumes. 13th European Nitrogen Fixation Conference (ENFC); ENFC: Stockholm, Sweden, 2018; p. 204. [Google Scholar]
- Aksenova, T.S.; Onishchuk, O.P.; Kurchak, O.N.; Igolkina, A.A.; Andronov, E.E.; Provorov, N.A. Positive selection in populations of nodule bacteria Rhizobium leguminosarum bv. Trifolii. In VII Congress of Vavilov Society of Genetics and Breeders on the 100th Anniversary of the Department of Genetics of Saint Petersburg State University, and Associate Symposia. Saint Petersburg; Saint Petersburg State University: Saint Petersburg, Russian, 2019; p. 662. [Google Scholar]
- Ovtsyna, A.O.; Rademaker, G.J.; Esser, E.; Weinman, J.; Rolfe, B.G.; Tikhonovich, I.A.; Lugtenberg, B.J.; Thomas-Oates, J.E.; Spaink, H.P. Comparison of characteristics of the nodX genes from various Rhizobium leguminosarum strains. Mol. Plant Microbe Interact. 1999, 12, 252–258. [Google Scholar] [CrossRef]
- Provorov, N.A.; Tikhonovich, I.A. Genetic resources for improving nitrogen fixation in legume-rhizobia symbiosis. Genet. Res. Crop Evol. 2003, 50, 89–99. [Google Scholar] [CrossRef]
- Provorov, N.A.; Vorobyov, N.I. Evolutionary Genetics of Plant-Microbe Symbioses (Agronomy Research and Developments); Nova Science Pub Inc.: New York, NY, USA, 2010; pp. 1–290. [Google Scholar]
- del Carmen Orozco-Mosqueda, M.; Altamirano-Hernandez, J.; Farias-Rodriguez, R.; Valencia-Cantero, E.; Santoyo, G. Homologous recombination and dynamics of rhizobial genomes. Res. Microbiol. 2009, 160, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Karasev, E.S.; Chizhevskaya, E.P.; Simarov, B.V.; Provorov, N.A.; Andronov, E.E. Comparative phylogenetic analysis of symbiotic genes of different nodule bacteria groups using the metatrees method. Agric. Biol. 2017, 52, 995–1003. [Google Scholar] [CrossRef]
- Vavilov, N.I. The origin, variation, immunity and breeding of cultivated plants. Chron. Bot. 1951, 13, 1–364. [Google Scholar] [CrossRef]
- Safronova, V.I.; Belimov, A.A.; Sazanova, A.L.; Chirak, E.R.; Verkhozina, A.V.; Kuznetsova, I.G.; Andronov, E.E.; Puhalsky, J.V.; Tikhonovich, I.A. Taxonomically Different Co-Microsymbionts of a Relict Legume, Oxytropis popoviana, Have Complementary Sets of Symbiotic Genes and Together Increase the Efficiency of Plant Nodulation. Mol. Plant Microbe Interact. 2018, 31, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapp, D.; Niehaus, K.; Quandt, J.; Muller, P.; Puhler, A. Cooperative Action of Rhizobium meliloti Nodulation and Infection Mutants during the Process of Forming Mixed Infected Alfalfa Nodules. Plant Cell 1990, 2, 139–151. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Region | Host Plant | Accession No. (GenBank) | Genome Size, Mb | No. of Contigs/Replicons | No. of Annotated Genes | GC% | Reference |
|---|---|---|---|---|---|---|---|---|
| Vaf-12 | North Ossetia, Russia | V. formosa | LVYU01000001-LVYU01000166—whole genome KT944070—Sym region only | 7.666 | 166 | 7543 | 60.70 | [32] |
| Vaf-10 | North Ossetia, Russia | V. formosa | CP016286-CP016293 | 8.568 | 8 | 8559 | 60.53 | Current work |
| Vaf-108 | Dagestan, Russia | V. formosa | CP018228-CP018236 | 8.447 | 9 | 8320 | 60.50 | Current work |
| TOM | Turkey | P. sativum Afg | AQUC01000001-AQUC01000006 | 7.358 | 6 | 7192 | 60.80 | [33] |
| 3841 | UK | P. sativum | AM236080-AM236086 | 7.751 | 7 | 7599 | 60.87 | [34] |
| 248 | UK | V. faba | ARRT01000001-ARRT01000007 | 7.289 | 7 | 7148 | 60.90 | [33] |
| WSM1481 | Greece | V. faba | AQUM01000001-AQUM01000006 | 7.556 | 6 | 7452 | 61.00 | [33] |
| COG Group | Function | Vaf-10 | Vaf-12 | Vaf-108 | 3841 |
|---|---|---|---|---|---|
| C | Energy production and conversion | 1 | 2 | 3 | 0 |
| E | Amino acid transport and metabolism | 8 | 14 | 14 | 0 |
| H | Coenzyme transport and metabolism | 1 | 2 | 1 | 0 |
| I | Lipid transport and metabolism | 2 | 2 | 2 | 0 |
| J | Translation, ribosomal structure and biogenesis | 1 | 1 | 1 | 0 |
| K | Transcription | 1 | 6 | 5 | 1 |
| L | Replication, recombination and repair | 2 | 1 | 0 | 1 |
| M | Cell wall/membrane/envelope biogenesis | 0 | 1 | 0 | 1 |
| O | Post-translational modification, protein turnover, and chaperones | 1 | 1 | 1 | 0 |
| P | Inorganic ion transport and metabolism | 4 | 3 | 2 | 0 |
| R | General function prediction only | 1 | 2 | 2 | 0 |
| S | Function unknown | 0 | 1 | 1 | 1 |
| T | Signal transduction mechanisms | 1 | 2 | 2 | 1 |
| V | Defense mechanisms | 2 | 2 | 2 | 0 |
| X | Mobile elements | 6 | 1 | 12 | 4 |
| Total | 31 | 41 | 48 | 9 |
| Group | Protein | Function | Sym plasmid | Chromosome | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vaf-10 | Vaf-12 | Vaf-108 | 3841 | Vaf-10 | Vaf-12 | Vaf-108 | 3841 | ||||
| COG0136 | E | Asd | Aspartate-semialdehyde dehydrogenase | + | + | + | + | + | + | + | |
| COG0334 | E | GdhA | NADP-specific glutamate dehydrogenase | + | + | + | |||||
| COG0410 | E | LivF | ABC-type branched-chain amino acid transport systems, ATPase component | + | + | + | + | + | + | ||
| COG0411 | E | LivG | ABC-type branched-chain amino acid transport systems, ATPase component | + | + | + | + | + | + | ||
| COG0527 | E | LysC | Aspartokinase | + | + | + | |||||
| COG0559 | E | LivH | Branched-chain amino acid ABC-type transport system, permease components | + | + | + | + | + | + | ||
| COG0683 | E | LivK | ABC-type branched-chain amino acid transport systems, periplasmic component | + | + | + | + | + | + | ||
| COG0747 | E | OppA | ABC-type dipeptide transport system, periplasmic component | + | + | + | + | + | + | + | |
| COG1982 | E | LdcC | arginine/lysine/ornithine decarboxylase | + | + | + | + | + | + | + | |
| COG4177 | E | LivM | ABC-type branched-chain amino acid transport system, permease component | + | + | + | + | + | + | ||
| COG0601 | EP | DppB | ABC-type dipeptide/oligopeptide/nickel transport systems, permease components | + | + | + | + | + | + | + | |
| COG1173 | EP | NikC | ABC-type dipeptide/oligopeptide/nickel transport systems, permease components | + | + | + | + | + | + | + | |
| COG0583 | K | LysR | Transcriptional regulator, LysR family | + | + | + | + | + | |||
| COG1508 | K | RpoN | DNA-directed RNA polymerase specialized sigma subunit, sigma54 homolog | + | + | + | + | + | |||
| COG1522 | K | Lrp | putative AsnC family transcriptional regulatory protein | + | + | + | + | + | |||
| COG1737 | K | RpiR | Transcriptional regulator, nylB upstream ORF | + | + | + | + | + | + | + | |
| COG4977 | K | AraC | Transcriptional regulator AraC family | + | + | + | + | + | + | ||
| COG1167 | KE | ARO8 | Transcriptional regulator, GntR family domain/Aspartate aminotransferase | + | + | + | + | + | + | ||
| COG2771 | K | RhiR | DNA-binding HTH domain-containing proteins | + | + | + | + | + | + | + | |
| COG3637 | M | RhiD | Opacity protein and related surface antigens | + | + | ||||||
| COG3916 | T | RhiI | N-acyl-L-homoserine lactone synthetase | + | + | ||||||
| COG4675 | S | RhiB | Microcystin-dependent protein | + | + | + | |||||
| not in COG | RhiA | + | + | + | |||||||
| not in COG | RhiC | + | + | + | |||||||
| nodT-nodO Distance | nodT Location | nodX | fixW | Sym Region Length | fixNOQP Copies | |
|---|---|---|---|---|---|---|
| Vaf-10 | not linked | Separate | + | + | 93 kb | 1 |
| Vaf-108 | 30 kb | Separate | + | + | 115 kb | 1 |
| Vaf-12 | 25 kb | Separate | + | + | 98 kb | 1 |
| TOM | 10 kb | Separate | + | + | 91 kb | 3 |
| 3841 | < 1 kb | Between nodO and nodN | – | – | 59 kb | 3 |
| 248 | < 1 kb | Between nodO and nodN | – | – | 57 kb | 2 |
| WSM1481 | < 1 kb | Between nodO and nodN | – | – | 54 kb | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chirak, E.R.; Kimeklis, A.K.; Karasev, E.S.; Kopat, V.V.; Safronova, V.I.; Belimov, A.A.; Aksenova, T.S.; Kabilov, M.R.; Provorov, N.A.; Andronov, E.E. Search for Ancestral Features in Genomes of Rhizobium leguminosarum bv. viciae Strains Isolated from the Relict Legume Vavilovia formosa. Genes 2019, 10, 990. https://doi.org/10.3390/genes10120990
Chirak ER, Kimeklis AK, Karasev ES, Kopat VV, Safronova VI, Belimov AA, Aksenova TS, Kabilov MR, Provorov NA, Andronov EE. Search for Ancestral Features in Genomes of Rhizobium leguminosarum bv. viciae Strains Isolated from the Relict Legume Vavilovia formosa. Genes. 2019; 10(12):990. https://doi.org/10.3390/genes10120990
Chicago/Turabian StyleChirak, Elizaveta R., Anastasiia K. Kimeklis, Evgenii S. Karasev, Vladimir V. Kopat, Vera I. Safronova, Andrey A. Belimov, Tatiana S. Aksenova, Marsel R. Kabilov, Nikolay A. Provorov, and Evgeny E. Andronov. 2019. "Search for Ancestral Features in Genomes of Rhizobium leguminosarum bv. viciae Strains Isolated from the Relict Legume Vavilovia formosa" Genes 10, no. 12: 990. https://doi.org/10.3390/genes10120990
APA StyleChirak, E. R., Kimeklis, A. K., Karasev, E. S., Kopat, V. V., Safronova, V. I., Belimov, A. A., Aksenova, T. S., Kabilov, M. R., Provorov, N. A., & Andronov, E. E. (2019). Search for Ancestral Features in Genomes of Rhizobium leguminosarum bv. viciae Strains Isolated from the Relict Legume Vavilovia formosa. Genes, 10(12), 990. https://doi.org/10.3390/genes10120990