Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome

 ,
, 


Abstract
:1. Introduction
2. Patient and Methods
2.1. Molecular Analyses
2.2. High-Frequency Ultrasonography and In Vivo Reflectance Confocal Microscopy
3. Results
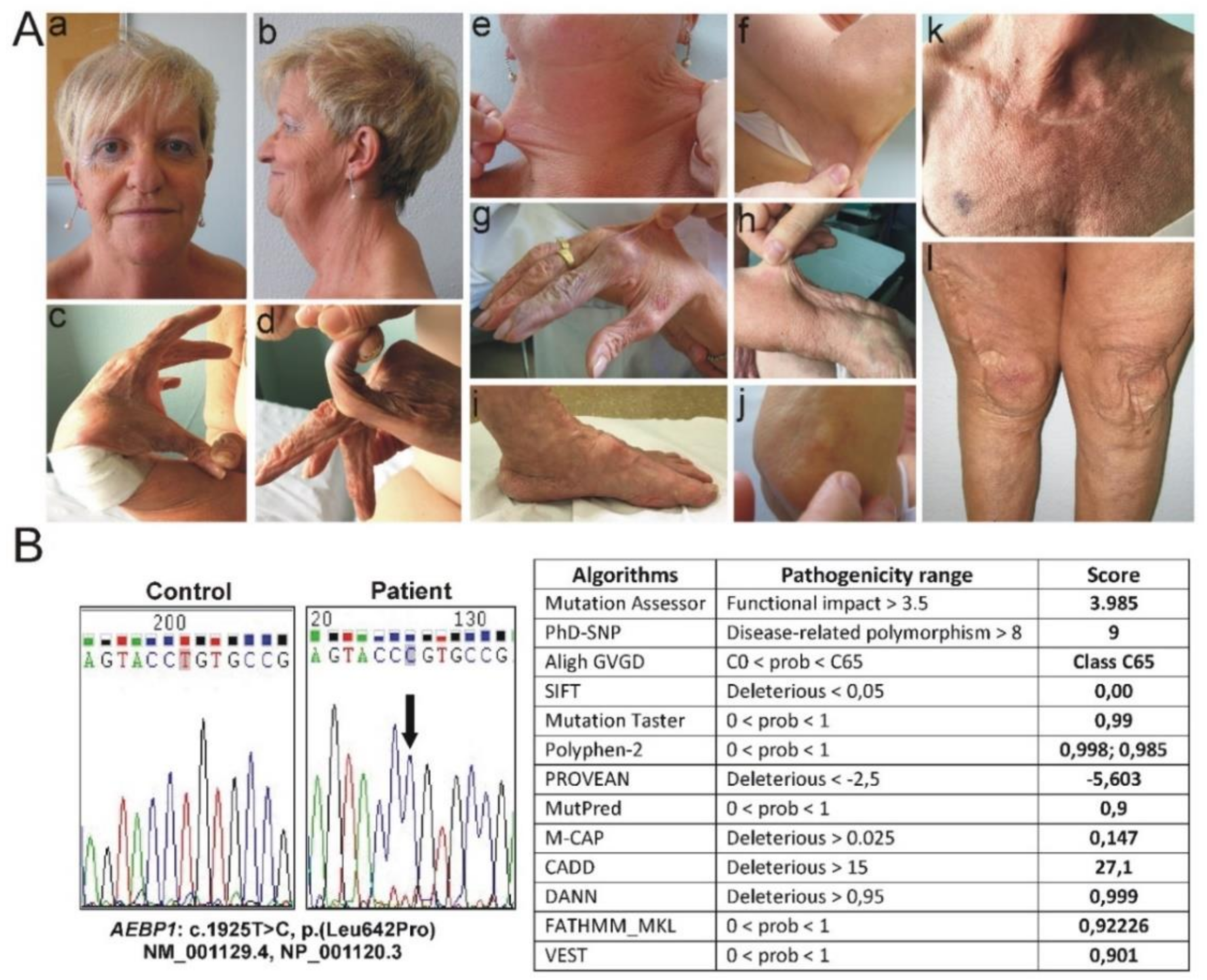
3.1. Clinical Findings
3.2. Molecular Findings
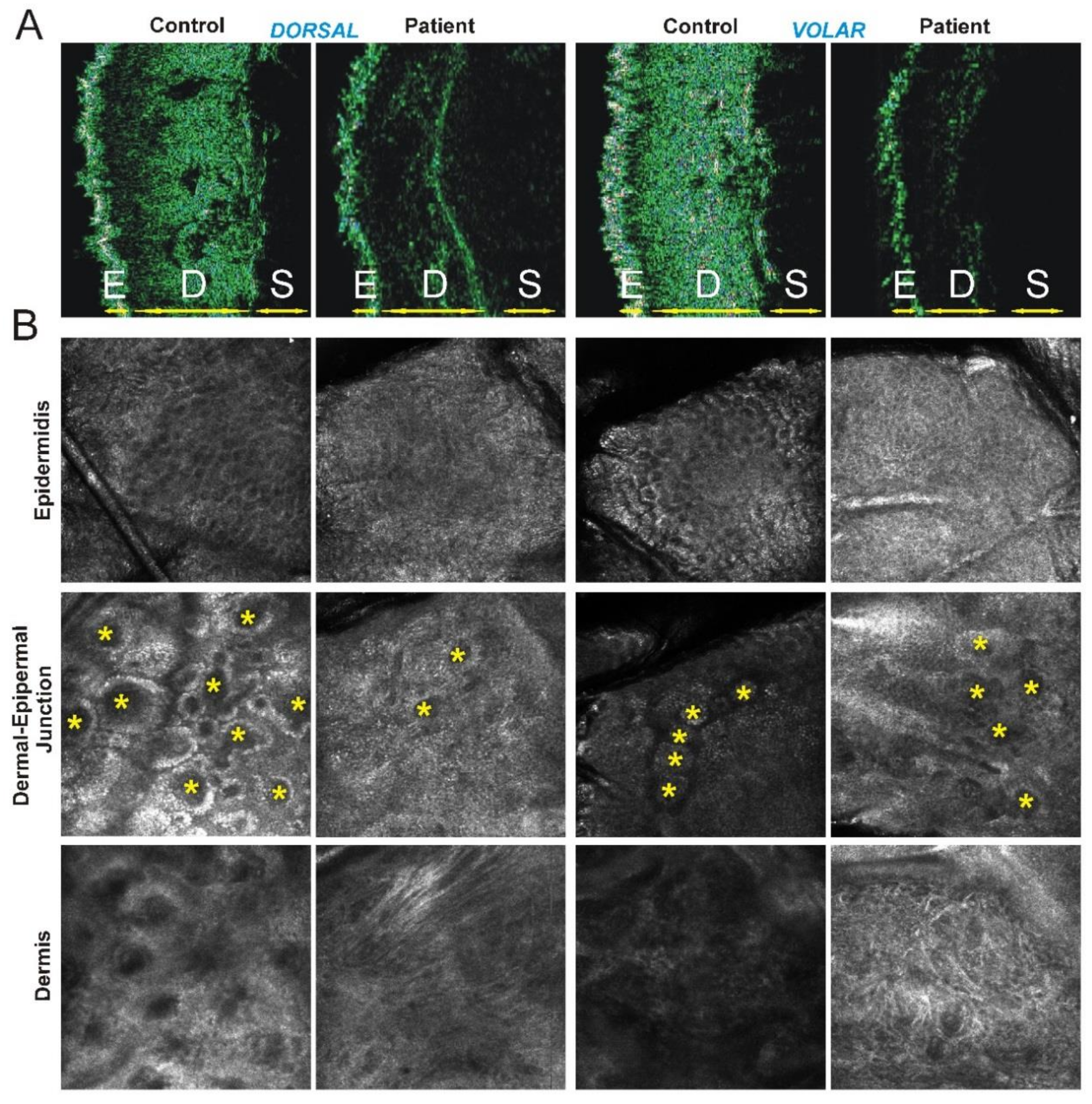
3.3. Instrumental Findings on Patient’s Skin
4. Discussion
5. Conclusions
Supplementary Materials
Author contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. Part C 2017, 75, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Ritelli, M.; Dordoni, C.; Venturini, M.; Chiarelli, N.; Quinzani, S.; Traversa, M.; Zoppi, N.; Vascellaro, A.; Wischmeijer, A.; Manfredini, E.; et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J. Rare Dis. 2013, 12, 8–58. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Venturini, M.; Ciaccio, C.; Morlino, S.; Chiarelli, N.; Zanca, A.; Calzavara-Pinton, P.; Zoppi, N.; Castori, M.; et al. Spectrum of mucocutaneous, ocular and facial features and delineation of novel presentations in 62 classical Ehlers-Danlos syndrome patients. Clin. Genet. 2017, 92, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Cinquina, V.; Venturini, M.; Ritelli, M. A classical Ehlers-Danlos syndrome family with incomplete presentation diagnosed by molecular testing. Eur. J. Med. Genet. 2018, 61, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Dordoni, C.; Doga, M.; Galderisi, F.; Venturini, M.; Calzavara-Pinton, P.; Maroldi, R.; Giustina, A.; Colombi, M. High prevalence of radiological vertebral fractures in adult patients with Ehlers-Danlos syndrome. Bone 2016, 84, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Borck, G.; Beighton, P.; Wilhelm, C.; Kohlhase, J.; Kubisch, C. Arterial rupture in classic Ehlers-Danlos syndrome with COL5A1 mutation. Am. J. Med. Genet. Part A 2010, 152, 2090–2093. [Google Scholar] [CrossRef]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Venturini, M.; Zanca, A.; Calzavara-Pinton, P.; Ritelli, M. Delineation of Ehlers-Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: Report on a three-generation family without cardiovascular events, and literature review. Am. J. Med. Genet. Part A 2017, 173, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. Part C 2015, 169, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. Part C 2017, 175, 48–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castori, M.; Dordoni, C.; Morlino, S.; Sperduti, I.; Ritelli, M.; Valiante, M.; Chiarelli, N.; Zanca, A.; Celletti, C.; Venturini, M.; et al. Spectrum of mucocutaneous manifestations in 277 patients with joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am. J. Med. Genet. Part C 2015, 169, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Hebebrand, M.; Vasileiou, G.; Krumbiegel, M.; Kraus, C.; Uebe, S.; Ekici, A.B.; Thiel, C.T.; Reis, A.; Popp, B. A biallelic truncating AEBP1 variant causes connective tissue disorder in two siblings. Am. J. Med. Genet. A 2018. [Google Scholar] [CrossRef]
- GnomAD Database. Available online: http://gnomad.broadinstitute.org/ (accessed on 7 January 2019).
- Dalgleish, R. The human collagen mutation database 1998. Nucleic Acids Res. 1998, 26, 253–255. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef]
- PhD-SNP Web Server. Available online: http://snps.biofold.org/phd-snp/phd-snp.html (accessed on 21 August 2018).
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Ning Lin, G.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 2017, 134981. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. 3), S3. [Google Scholar] [CrossRef]
- Polańska, A.; Dańczak-Pazdrowska, A.; Jałowska, M.; Żaba, R.; Adamski, Z. Current applications of high-frequency ultrasonography in dermatology. Postepy Dermatol. Alergol. 2017, 34, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calzavara-Pinton, P.; Longo, C.; Venturini, M.; Sala, R.; Pellacani, G. Reflectance confocal microscopy for in vivo skin imaging. Photochem. Photobiol. 2008, 84, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Rajadhyaksha, M.; González, S.; Zavislan, J.M.; Anderson, R.R.; Webb, R.H. In vivo confocal scanning laser microscopy of human skin II: Advances in instrumentation and comparison with histology. J. Invest. Dermatol. 1999, 113, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, S.J.; Theodorou, D.J.; Kakitsubata, Y.; Adams, J.E. Low bone mass in Ehlers–Danlos syndrome. Intern Med. 2012, 51, 3225–3226. [Google Scholar] [CrossRef] [PubMed]
- Reznik, S.E.; Fricker, L.D. Carboxypeptidases from A to Z: Implications in embryonic development and Wnt binding. Cell Mol. Life Sci. 2001, 58, 1790–1804. [Google Scholar] [CrossRef] [PubMed]
- Layne, M.D.; Yet, S.F.; Maemura, K.; Hsieh, C.M.; Bernfield, M.; Perrella, M.A.; Lee, M.E. Impaired abdominal wall development and deficient wound healing in mice lacking aortic carboxypeptidase-like protein. Mol. Cell Biol. 2001, 21, 5256–5261. [Google Scholar] [CrossRef] [PubMed]
- Ith, B.; Wei, J.; Yet, S.F.; Perrella, M.A.; Layne, M.D. Aortic carboxypeptidase-like protein is expressed in collagen-rich tissues during mouse embryonic development. Gene Expr. Patterns 2005, 5, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Layne, M.D.; Endege, W.O.; Jain, M.K.; Yet, S.F.; Hsieh, C.M.; Chin, M.T.; Perrella, M.A.; Blanar, M.A.; Haber, E.; Lee, M.E. Aortic carboxypeptidase-like protein, a novel protein with discoidin and carboxypeptidase-like domains, is up-regulated during vascular smooth muscle cell differentiation. J. Biol. Chem. 1998, 273, 15654–15660. [Google Scholar] [CrossRef] [PubMed]
- Layne, M.D.; Yet, S.F.; Maemura, K.; Hsieh, C.M.; Liu, X.; Ith, B.; Lee, M.E.; Perrella, M.A. Characterization of the mouse aortic carboxypeptidase-like protein promoter reveals activity in differentiated and dedifferentiated vascular smooth muscle cells. Circ. Res. 2002, 90, 728–736. [Google Scholar] [CrossRef]
- Schissel, S.L.; Dunsmore, S.E.; Liu, X.; Shine, R.W.; Perrella, M.A.; Layne, M.D. Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am. J. Pathol. 2009, 174, 818–828. [Google Scholar] [CrossRef]
- Tumelty, K.E.; Smith, B.D.; Nugent, M.A.; Layne, M.D. Aortic carboxypeptidase-like protein (ACLP) enhances lung myofibroblast differentiation through transforming growth factor β receptor-dependent and -independent pathways. J. Biol. Chem. 2014, 289, 2526–2536. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Dorsal Forearm | Volar Forearm | |||
|---|---|---|---|---|
| Patient | Controls (mean ± SD) | Patient | Controls (mean ± SD) | |
| Epidermal thickness (µM) | 172 | 121 ± 22 | 141 | 102 ± 12 |
| Dermal thickness (µM) | 570 | 1108 ± 320 | 289 | 983 ± 205 |
| Citation | Present patient | P1* | P2* | P3* | P4* | P5* | P6* |
|---|---|---|---|---|---|---|---|
| Sex | female | male | male | female | male | female | male |
| Ethnicity | white | white | white | Middle Eastern | Middle Eastern | white | white |
| Age at evaluation | 53y | 35y | 33y | 12y | 24y | 39y | 38y |
| AEBP1 variant(s) (NM_001129.4) | c.1925T>C homozygous | c.1470del, c.1743C>A compound heterozygous | c.1320_1326del homozygous | c.1630+1G>A (r.1609_1630del) homozygous | c.1630+1G>A (r.1609_1630del) homozygous | c.917dup homozygous | c.917dup homozygous |
| Protein change (NP_001120.3) | p.(Leu642Pro) | p.(Asn490_Met495delins40), p.(Cys581*) | p.(Arg440Serfs*3) | p.(Val537Leufs*31) | p.(Val537Leufs*31) | p.(Tyr306*) | p.(Tyr306*) |
| Joint hypermobility (BS) | + (5/9) | + (8/9) | + (8/9) | + (8/9) | + (NA) | + (6/9) | + (2/9) |
| Dislocations/ Subluxations | left ankle, knees, shoulders, elbows | hip, right distal radioulnar joint | hip (congenital), shoulders | hip, knees, ankles shoulders, interphalangeal joints | hips, knees and ankles | wrist, mandibular and distal radioulnar joints | ankles, knees, clavicula |
| Foot deformities | pes planus, hallux valgus | pes planus, hallux valgus, hammer toes | pes planus, hallux valgus, hammer toes | pes planus, hallux valgus, hammer toes | pes planus, hallux valgus, toe deformities | pes planus, hallux valgus, sandal gap | hindfoot deformity, sandal gap |
| Extensive skin hyperextensibility | + | + | + | + | + | + | + |
| Delayed would healing (abnormal scarring) | + (widened atrophic scars) | + (widened atrophic scars) | + (widened atrophic scars, keloids) | + (widened atrophic scars, keloids) | + (widened atrophic scars) | + (widened atrophic scars) | + (widened atrophic scars) |
| Redundant skin | + old-aging appearance | + old-aging appearance | + | + | + | + old-aging appearance | + old-aging appearance |
| Easy bruising | + | + | + | + | NA | + | + |
| Prominent chest superficial veins | - | NA | + | NA | NA | + | + |
| Hernia | umbilical surgically treated | - | large ventral surgical hernia | umbilical, ventral, inguinal | NA | + | - |
| Genitourinary abnormalities | - | cryptorchidism surgically corrected | - | - | - | - | cryptorchidism surgically corrected |
| Gastrointestinal abnormalities | - | motility issues | bowel rupture | - | - | NA | NA |
| Vascular abnormalities | peripheral artery disease, varicose veins | MVP | MVP, mildly dilated aortic root, bilateral carotids stenosis, aortic dilation requiring surgery | - | - | MVP, circular pericardial effusion | varicose veins |
| Dentition | Pyorrhea, complete dental loss at age 14 | retention of a single baby tooth | - | abnormal dental alignment | abnormal dental alignment | - | - |
| Citation | Present patient | P1* | P2* | P3* | P4* | P5* | P6* |
| Sex | female | male | male | female | male | female | male |
| Ethnicity | white | white | white | Middle Eastern | Middle Eastern | white | white |
| Age at evaluation | 53y | 35y | 33y | 12y | 24y | 39y | 38y |
| Facial dysmorphisms | high palate, elongated uvula | - | micrognathia | bilateral ptosis webbed neck, sagged cheeks large ears, narrow palate | bilateral ptosis webbed neck, sagged cheeks large ears, narrow palate | - | - |
| Skeletal anomalies (MRI findings) | femoral osteopenia, T10 vertebral deformity, scoliosis, lumbar spine rectilinization with marked degenerative arthritis | severe osteopenia of hips (mild disc bulging at the C4-5 and C7-T1 levels) | hip replacement for severe osteopenia, upper thoracic scoliosis with degenerative disease and facet arthrosis of spine (empty sella) | skull with ‘copper beaten’ appearance, severe osteopenia, narrowing of the interpedicular distance of the lumbar spine distally, short and squared iliac bones, remodeled long bones of the lower extremities | severe osteopenia | progressive kyphosis, scoliosis, arachnodactyly, positive wrist and thumb signs, degeneration of the discus ulnaris orthopedically treated | kyphoscoliosis, arachnodactyly, positive wrist and thumb signs, mild pectus excavatum |
| Other | hypotonia, delayed motor development, multiple papules (diffuse PoC-like dermatitis, alopecia, patellar instability surgically treated, rotator cuff disease surgically treated, epitrochleitis, subacromial shoulder impingement, hypotrophy of the scapular girdle, gonarthrosis, chronic fatigue, spheroids, piezogenic papules, myopia | delays in walking and acquisition of fine motor skills, impaired temperature sensation, keratoconjunctivitis sicca, piezogenic papules | elbow bursitis, piezogenic papules, sacral dimple, hypertriglyceridemia | hypotonia, diabetes mellitus, cellulitis | NA | alopecia, skin striae | strabismus surgically treated, myopia, astigmatism |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritelli, M.; Cinquina, V.; Venturini, M.; Pezzaioli, L.; Formenti, A.M.; Chiarelli, N.; Colombi, M. Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes 2019, 10, 135. https://doi.org/10.3390/genes10020135
Ritelli M, Cinquina V, Venturini M, Pezzaioli L, Formenti AM, Chiarelli N, Colombi M. Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes. 2019; 10(2):135. https://doi.org/10.3390/genes10020135
Chicago/Turabian StyleRitelli, Marco, Valeria Cinquina, Marina Venturini, Letizia Pezzaioli, Anna Maria Formenti, Nicola Chiarelli, and Marina Colombi. 2019. "Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome" Genes 10, no. 2: 135. https://doi.org/10.3390/genes10020135
APA StyleRitelli, M., Cinquina, V., Venturini, M., Pezzaioli, L., Formenti, A. M., Chiarelli, N., & Colombi, M. (2019). Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes, 10(2), 135. https://doi.org/10.3390/genes10020135