Identifying Selection Signatures for Backfat Thickness in Yorkshire Pigs Highlights New Regions Affecting Fat Metabolism



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Animals and Phenotype
2.3. Genotype and Quality Control
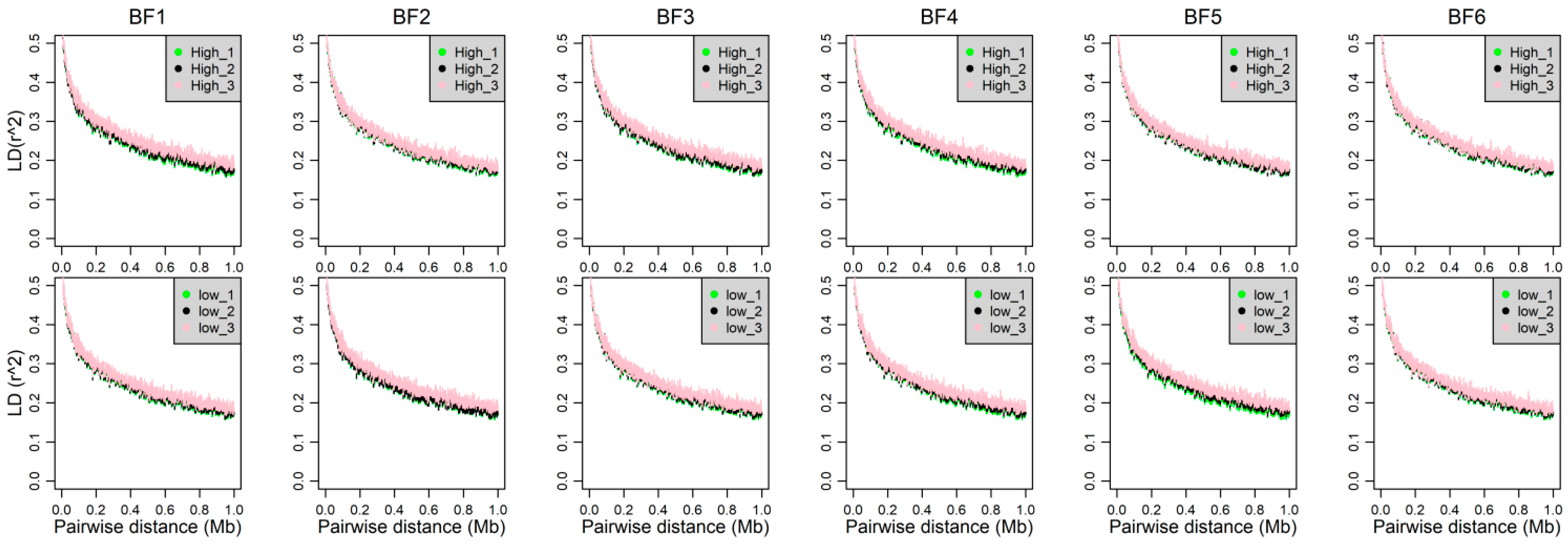
2.4. Linkage Disequilibrium, Allele Frequency and Heterozygosity
2.5. Detecting Postive Selection Signatures by iHS
2.6. Identification of Trait-Specific Selection Signatures
2.7. Functional Annotation for Trait-Specific Selection Signatures
3. Results
3.1. Qualified SNPs and Animals in the Analysis
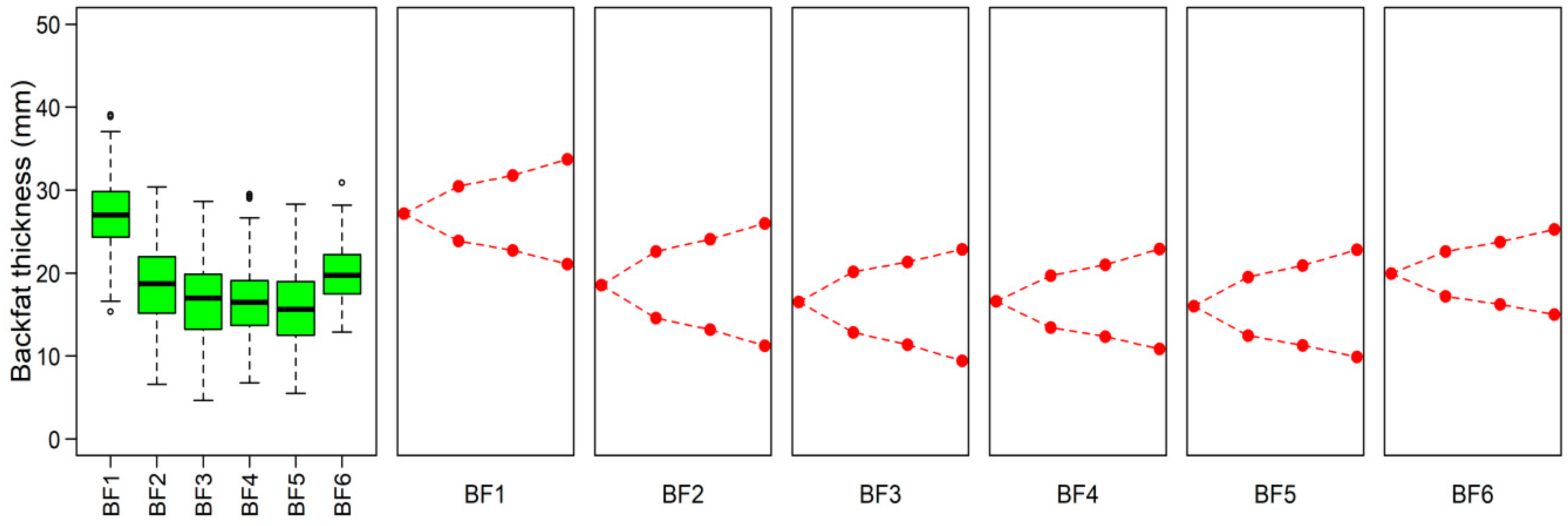
3.2. The summary of Phenotype
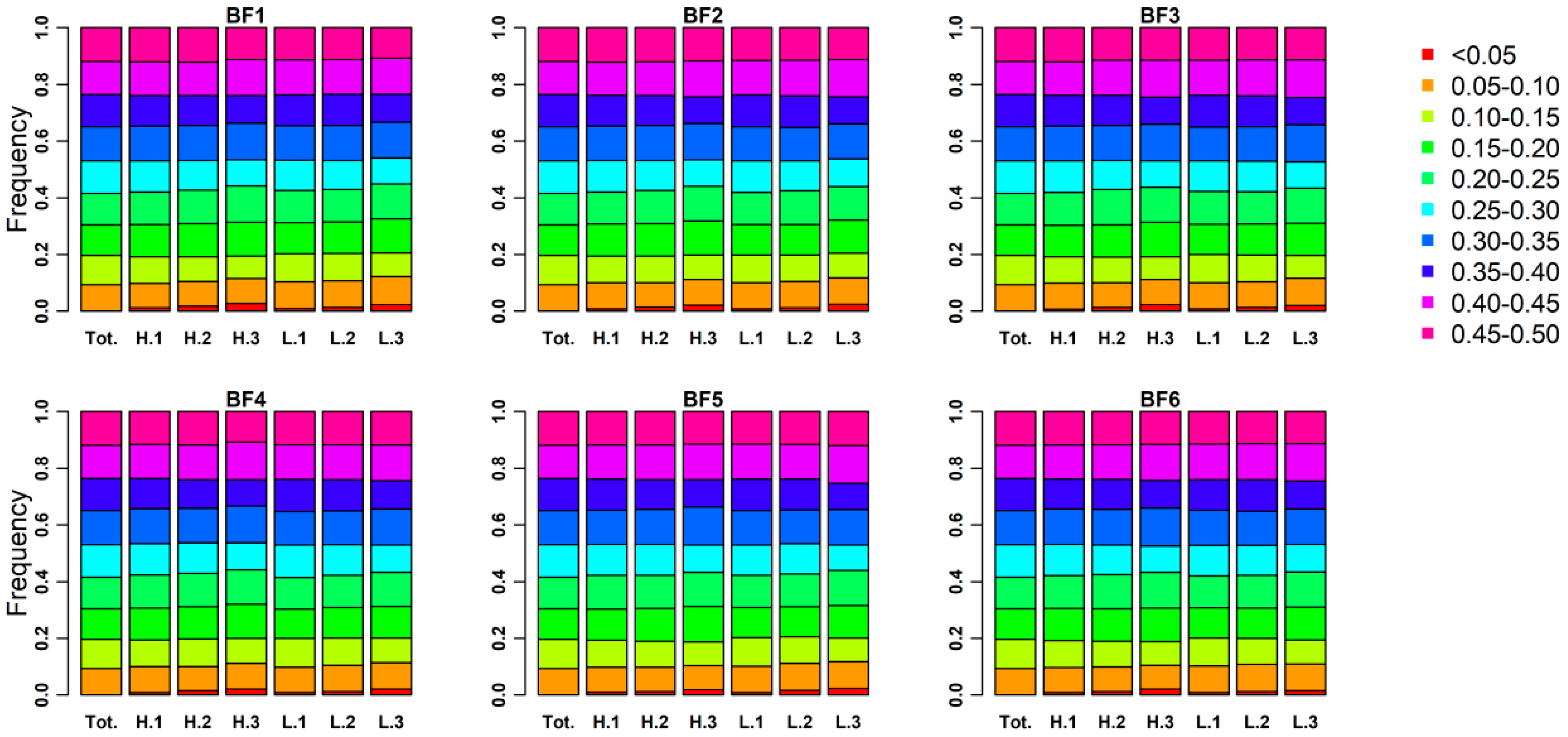
3.3. Genomic Characters among Three Phenotypic Gradient Differential Population Pairs
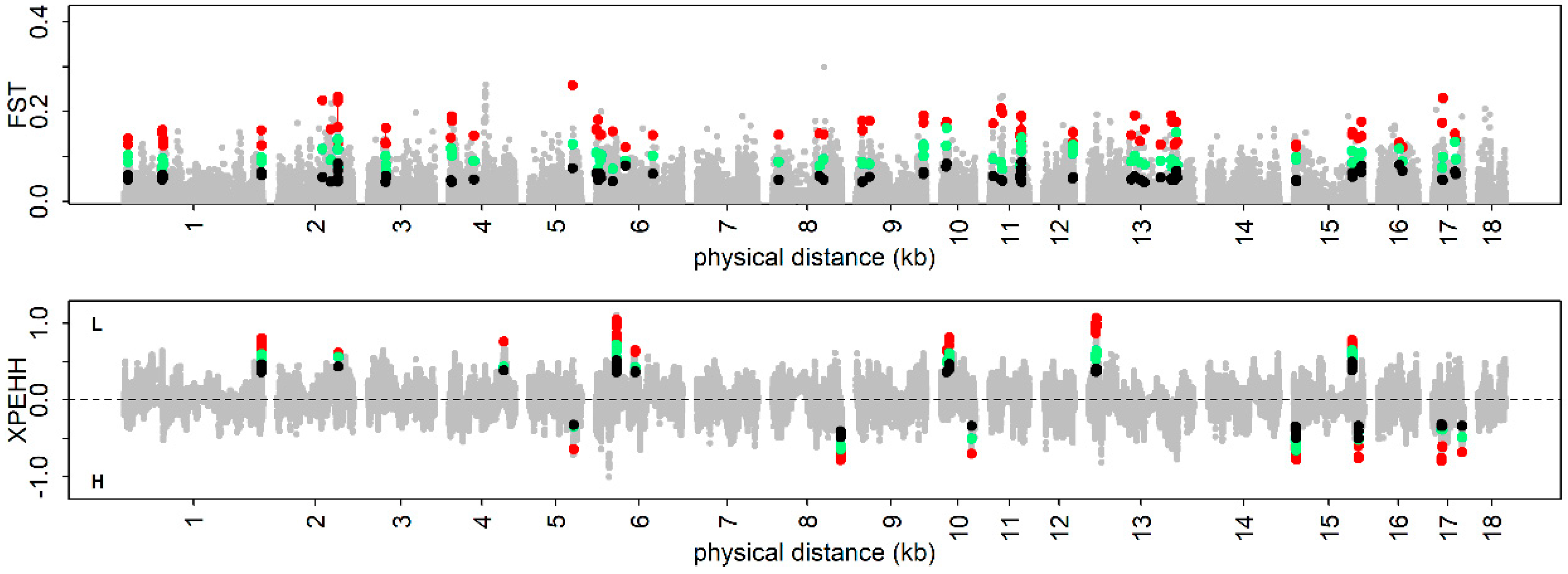
3.4. The Positive Selection Signatures
3.5. Trait-Specific Selection Signatures
3.6. Candidate Genes at the Identified Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bosi, P.; Russo, V. The production of the heavy pig for high quality processed products. Ital. J. Anim. Sci. 2010, 3, 309–321. [Google Scholar] [CrossRef]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calo, D.G.; Scotti, E.; Martelli, P.L.; Buttazzoni, L.; Casadio, R.; Russo, V. A genome wide association study for backfat thickness in Italian Large White pigs highlights new regions affecting fat deposition including neuronal genes. BMC Genomics 2012, 13, 583. [Google Scholar] [CrossRef]
- Guo, Y.M.; Qiu, H.Q.; Xiao, S.J.; Wu, Z.F.; Yang, M.; Yang, J.; Ren, J.; Huang, L.S. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J. Appl. Genet. 2017, 58, 499–508. [Google Scholar] [CrossRef]
- Jiang, Y.; Tang, S.; Wang, C.; Wang, Y.; Qin, Y.; Wang, Y.; Zhang, J.; Song, H.; Mi, S.; Yu, F.; et al. A genome-wide association study of growth and fatness traits in two pig populations with different genetic backgrounds. J. Anim. Sci. 2018, 96, 806–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eusebi, P.G.; Gonzalez-Prendes, R.; Quintanilla, R.; Tibau, J.; Cardoso, T.F.; Clop, A.; Amills, M. A genome-wide association analysis for carcass traits in a commercial Duroc pig population. Anim. Genet. 2017, 48, 466–469. [Google Scholar] [CrossRef]
- Edea, Z.; Hong, J.K.; Jung, J.H.; Kim, D.W.; Kim, Y.M.; Kim, E.S.; Shin, S.S.; Jung, Y.C.; Kim, K.S. Detecting selection signatures between Duroc and Duroc synthetic pig populations using high-density SNP chip. Anim. Genet. 2017, 48, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ding, X.; Qanbari, S.; Weigend, S.; Zhang, Q.; Simianer, H. Properties of different selection signature statistics and a new strategy for combining them. Heredity 2015, 115, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Ma, Y.L.; Zhang, S.X.; Zhang, K.L.; Fang, C.C.; Xie, S.S.; Du, X.Y.; Li, X.Y.; Ni, D.B.; Zhao, S.H. Genomic Analysis To Identify Signatures of Artificial Selection and Loci Associated with Important Economic Traits in Duroc Pigs. G3-Genes Genomes Genet. 2018, 8, 3617–3625. [Google Scholar] [CrossRef]
- Ma, Y.L.; Wei, J.L.; Zhang, Q.; Chen, L.; Wang, J.Y.; Liu, J.F.; Ding, X.D. A Genome Scan for Selection Signatures in Pigs. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.Q.; Pritchard, J.K. A map of recent positive selection in the human genome. PloS Biol. 2007, 5, e147. [Google Scholar] [CrossRef]
- Gautier, M.; Vitalis, R. rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 2012, 28, 1176–1177. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.H.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Qanbari, S.; Pausch, H.; Jansen, S.; Somel, M.; Strom, T.M.; Fries, R.; Nielsen, R.; Simianer, H. Classic Selective Sweeps Revealed by Massive Sequencing in Cattle. PLOS Genet. 2014, 10, e1004148. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.Y.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38. [Google Scholar] [CrossRef]
- Beaslas, O.; Metso, J.; Nissila, E.; Laurila, P.P.; Kaiharju, E.; Batchu, K.C.; Kaipiainen, L.; Mayranpaa, M.I.; Yan, D.; Gylling, H.; et al. Osbpl8 Deficiency in Mouse Causes an Elevation of High-Density Lipoproteins and Gender-Specific Alterations of Lipid Metabolism. PLoS ONE 2013, 8, e58856. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Dreier, J.L.; Ellis, J.M.; Allende, M.L.; Kalkofen, D.N.; Sanders, K.M.; Bielawski, J.; Bielawska, A.; Hannun, Y.A.; Proia, R.L. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J. Biol. Chem. 2006, 281, 7324–7331. [Google Scholar] [CrossRef] [PubMed]
- Pescador, N.; Villar, D.; Cifuentes, D.; Garcia-Rocha, M.; Ortiz-Barahona, A.; Vazquez, S.; Ordonez, A.; Cuevas, Y.; Saez-Morales, D.; Garcia-Bermejo, M.L.; et al. Hypoxia Promotes Glycogen Accumulation through Hypoxia Inducible Factor (HIF)-Mediated Induction of Glycogen Synthase 1. PLoS ONE 2010, 5, e9644. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARγ2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef] [Green Version]
- Bernard, F.; Moreau-Fauvarque, C.; Heitz-Marchaland, C.; Zagar, Y.; Dumas, L.; Fouquet, S.; Lee, X.H.; Shao, Z.H.; Mi, S.; Chedotal, A. Role of transmembrane semaphorin Sema6A in oligodendrocyte differentiation and myelination. Glia 2012, 60, 1590–1604. [Google Scholar] [CrossRef]
- Suzuki, K.; Ishida, M.; Kadowaki, H.; Shibata, T.; Uchida, H.; Nishida, A. Genetic correlations among fatty acid compositions in different sites of fat tissues, meat production, and meat quality traits in Duroc pigs. J. Anim. Sci. 2006, 84, 2026–2034. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Sanchez, J.; Amills, M.; Pena, R.N.; Mercade, A.; Manunza, A.; Quintanilla, R. Genomic architecture of heritability and genetic correlations for intramuscular and back fat contents in Duroc pigs. J. Anim. Sci. 2013, 91, 623–632. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr. | Position (bp) 1 | XPEHH (FST) Scores 2 | p-Values|iHS| | Genes 3 | Trait | MGI Phenotype |
|---|---|---|---|---|---|---|
| 5 | 39545743–39744655 | 0.28 < 0.39 < 0.48 | - | OSBPL8 | Low, BF4 | MP:0001552_increased circulating triglyceride level |
| 13 | 158574324–158574542 | −0.31 > −0.34 > −0.57 | - | ENSSSCG00000039018 (Tmem167) | High, BF6 | MP:0002644_decreased circulating triglyceride level |
| 7 | 114035383–114541592 | 0.29 < 0.38 < 0.69 | - | RIN3 | Low, BF4 | MP:0005560_decreased circulating glucose level |
| 17 | 22635204–22723444 | −0.32 > −0.37 > −0.64 | - | ENSSSCG00000030112 (Macrod2) | High, BF2 | MP:0013279_increased fasted circulating glucose level |
| 5 | 46244283–46274049 | 0.32 < 0.37 < 0.54 | 0.005 | SMCO2 | Low, BF4 | MP:0003960_increased lean body mass |
| 1 | 270911878–270912282 | 0.37 < 0.50 < 0.68 | 0.025 | QRFP | Low, BF1 | MP:0005375_adipose tissue phenotype MP:0001363_increased anxiety-related response |
| 14 | 99025306–99129155 | (0.03 < 0.06 < 0.11) | 0.002 | ENSSSCG00000010432 (ASAH2) | -, BF4 | MP:0001547_abnormal lipid level |
| 9 | 39864135–39916486 | (0.03 < 0.07 < 0.13) | 0.005 | ENSSSCG00000015039 (BCO2) | -, BF2 | MP:0000003_abnormal adipose tissue morphology |
| 1 | 270761674–270906709 | 0.36 < 0.49 < 0.67 | 0.025 | ABL1 | Low, BF1 | MP:0000598_abnormal liver morphology MP:0000607_abnormal hepatocyte morphology |
| 13 | 173634661–173910739 | −0.34 > −0.47 > −0.50, −0.31 > −0.44 > −0.80 | 0.032 | GBE1 | High, BF3, BF6 | MP:0000255_vasculature congestion MP:0005370_liver/biliary system phenotype |
| 1 | 26473935–26489776 | 0.33 < 0.50 < 0.13, 0.31 < 0.36 < 0.98 | 0.002 | TNFAIP3 | Low, BF5, BF6 | MP:0001845_abnormal inflammatory response |
| 13 | 158665077–158729282 | −0.31 > -0.34 > −0.57 | - | TBC1D23 | High, BF6 | MP:0001846_increased inflammatory response |
| 17 | 58998981–59055340 | −0.34 > −0.48 > −0.68 | 0.006 | GNAS | High, BF1 | MP:0001845_abnormal inflammatory response |
| 13 | 175980910–176479481 | −0.30 > −0.40 > −0.56 | 0.032 | ROBO1; | High, BF6 | MP:0005381_digestive/alimentary phenotype |
| 13 | 14754011–14785502 | 0.38 < 0.58 < 0.95 | - | AZI2 | Low, BF1 | MP:0002376_abnormal dendritic cell physiology |
| 2 | 120559078–120687331 | 0.44 < 0.56 < 0.62 | 0.021 | SEMA6A | Low, BF1 | MP:0000783_abnormal forebrain morphology |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, H.; Zhang, S.; Zhang, K.; Zhan, H.; Peng, X.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Identifying Selection Signatures for Backfat Thickness in Yorkshire Pigs Highlights New Regions Affecting Fat Metabolism. Genes 2019, 10, 254. https://doi.org/10.3390/genes10040254
Ma H, Zhang S, Zhang K, Zhan H, Peng X, Xie S, Li X, Zhao S, Ma Y. Identifying Selection Signatures for Backfat Thickness in Yorkshire Pigs Highlights New Regions Affecting Fat Metabolism. Genes. 2019; 10(4):254. https://doi.org/10.3390/genes10040254
Chicago/Turabian StyleMa, Haoran, Saixian Zhang, Kaili Zhang, Huiwen Zhan, Xia Peng, Shengsong Xie, Xinyun Li, Shuhong Zhao, and Yunlong Ma. 2019. "Identifying Selection Signatures for Backfat Thickness in Yorkshire Pigs Highlights New Regions Affecting Fat Metabolism" Genes 10, no. 4: 254. https://doi.org/10.3390/genes10040254
APA StyleMa, H., Zhang, S., Zhang, K., Zhan, H., Peng, X., Xie, S., Li, X., Zhao, S., & Ma, Y. (2019). Identifying Selection Signatures for Backfat Thickness in Yorkshire Pigs Highlights New Regions Affecting Fat Metabolism. Genes, 10(4), 254. https://doi.org/10.3390/genes10040254