The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
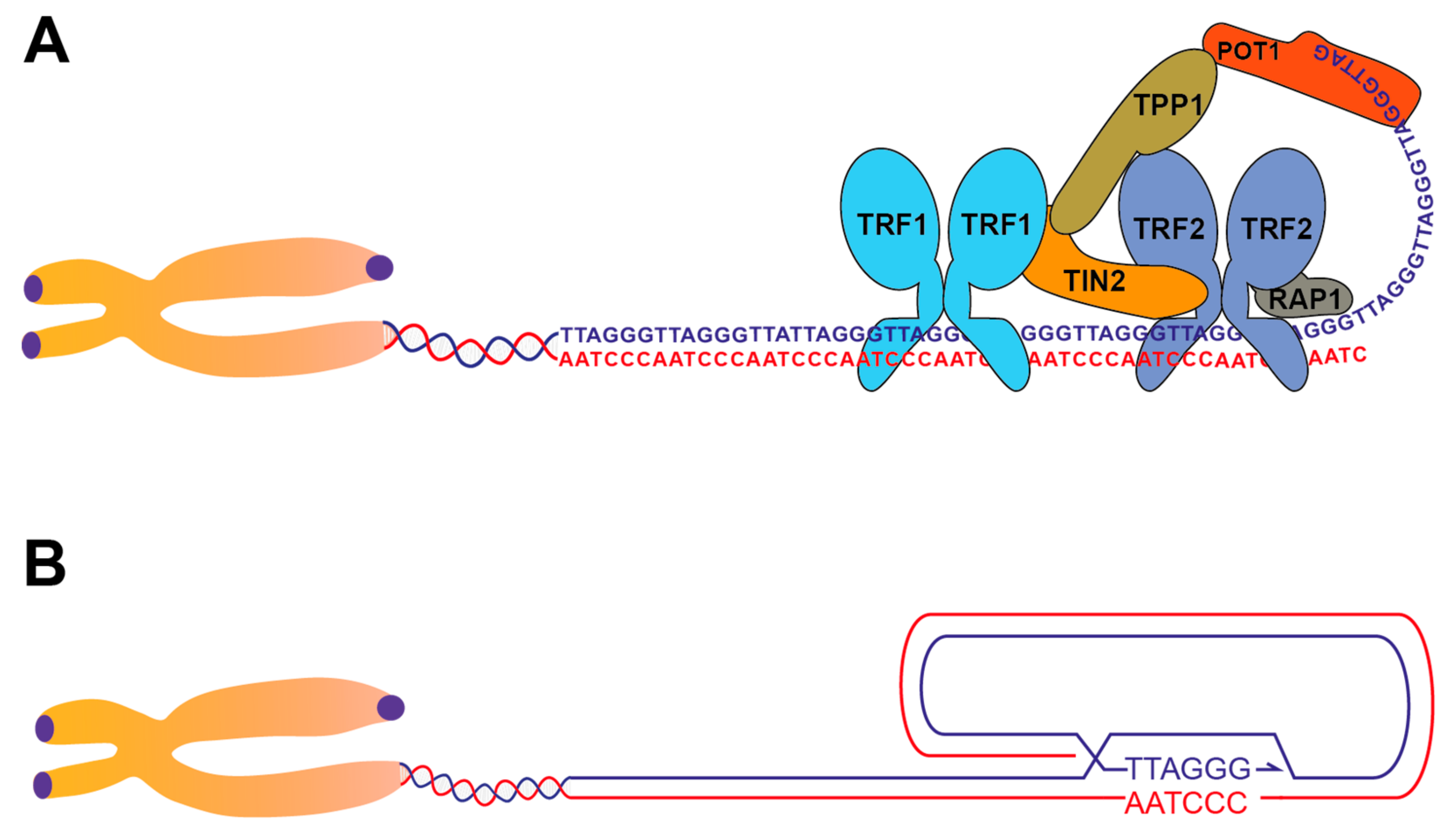
:1. Telomere Structure and End-Protection Functions
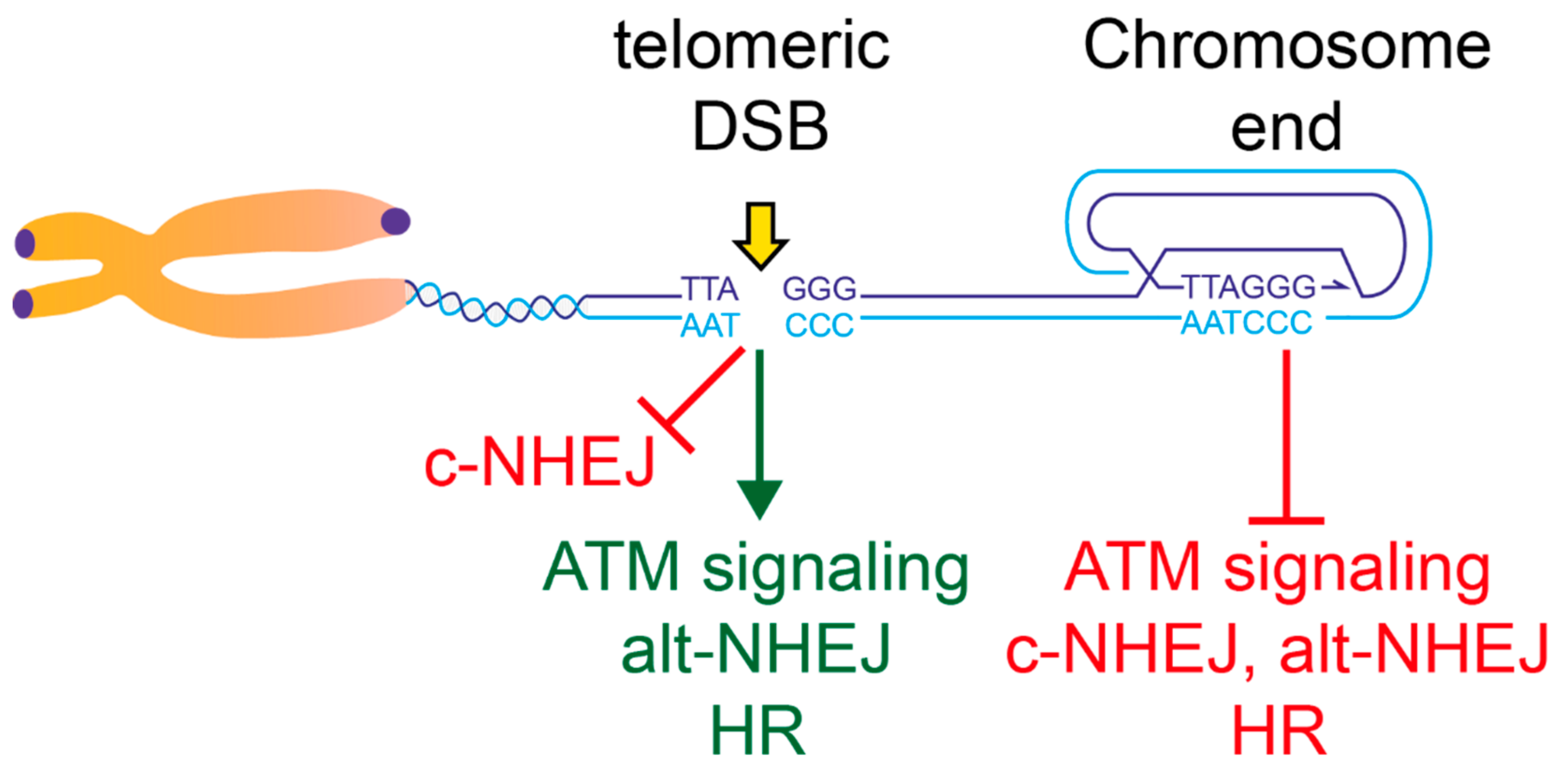
2. End-Protection vs. the Response to DNA Damage Inside the Telomeric Repeats
3. ATM Activation at Damaged Telomeres
4. C-NHEJ Repression at Telomeres
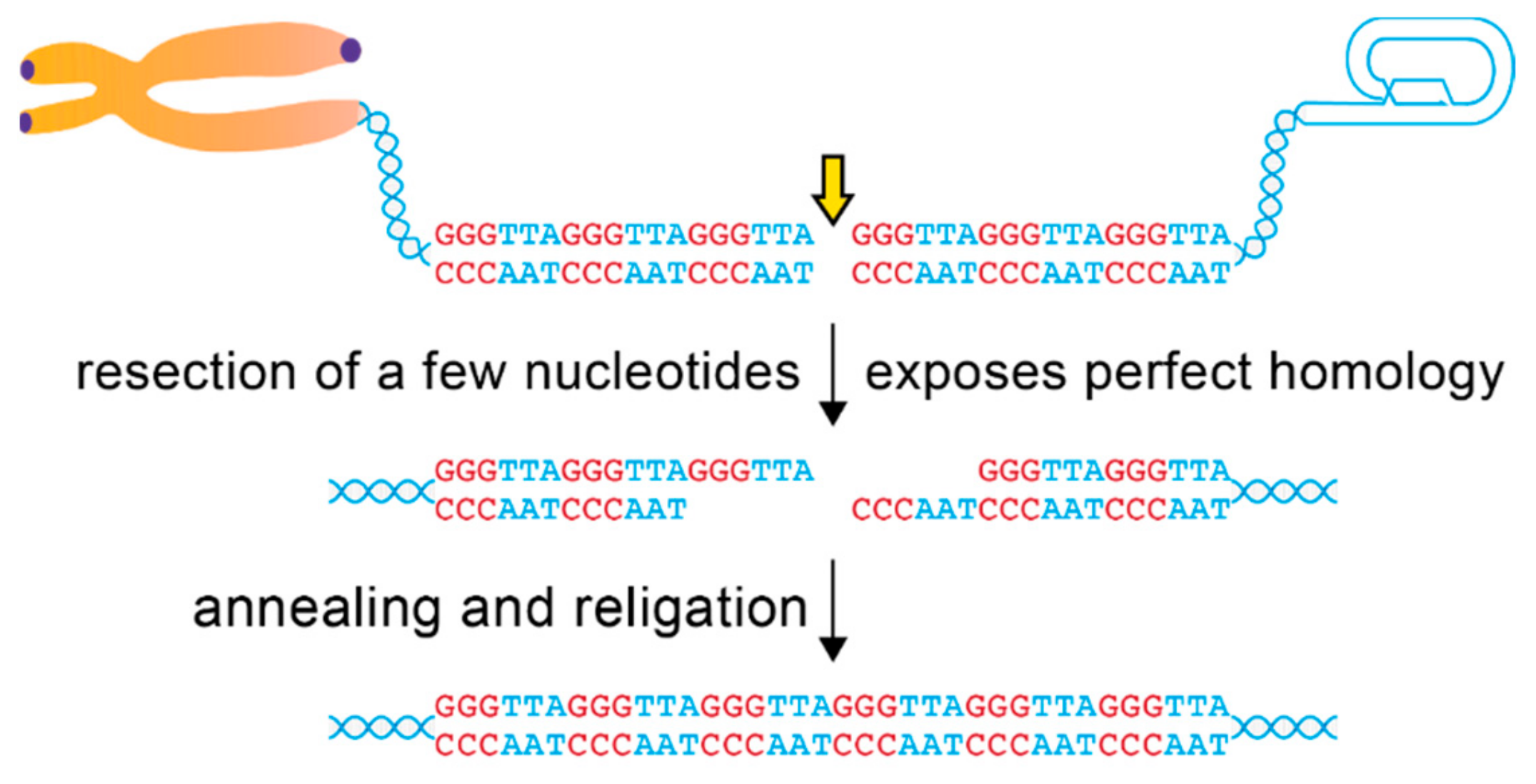
5. Alt-NHEJ for the Repair of Telomeric DSBs
6. Homologous Recombination during Telomere Replication
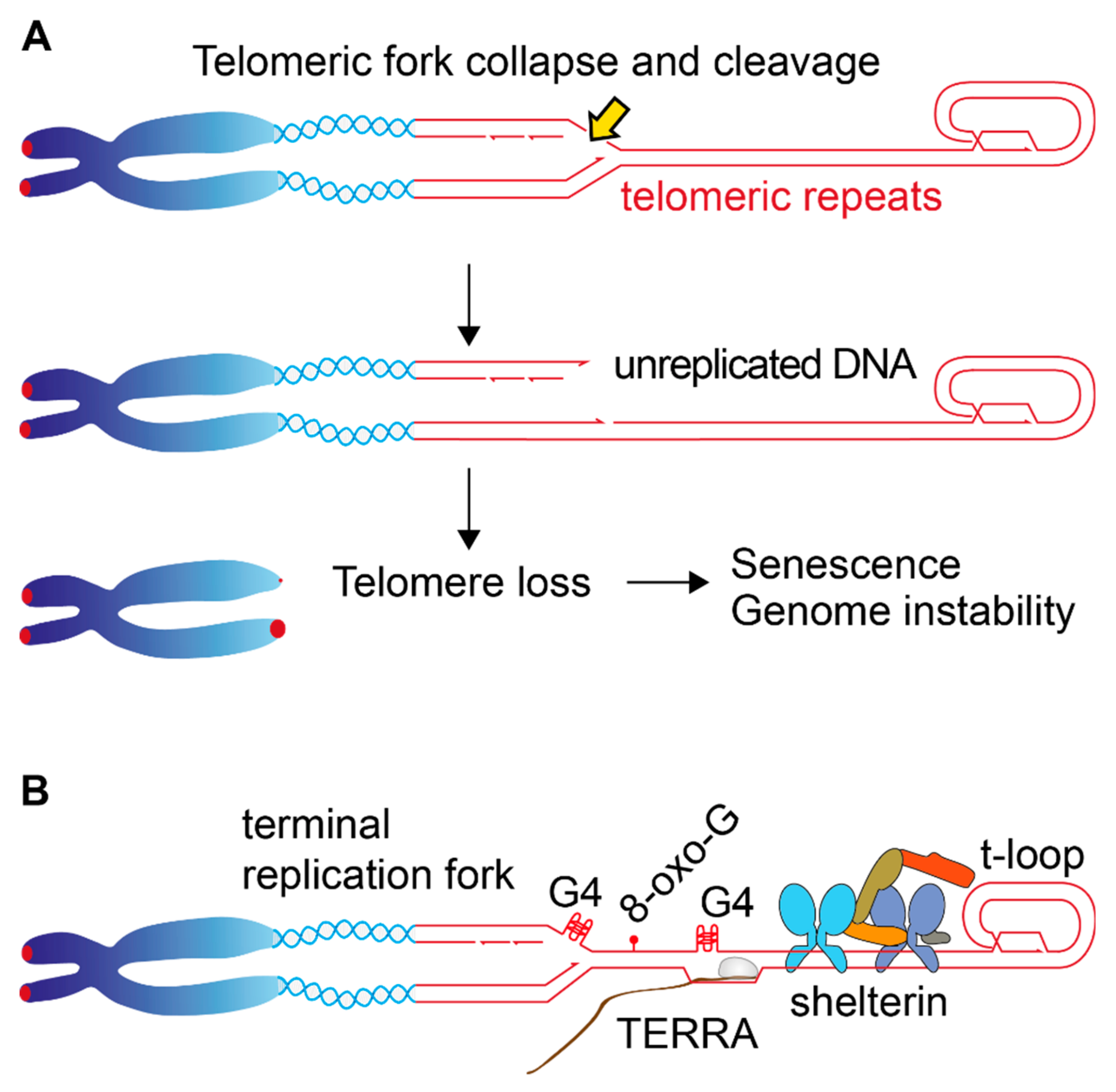
7. Telomere Loss, Beyond the End-Replication Problem
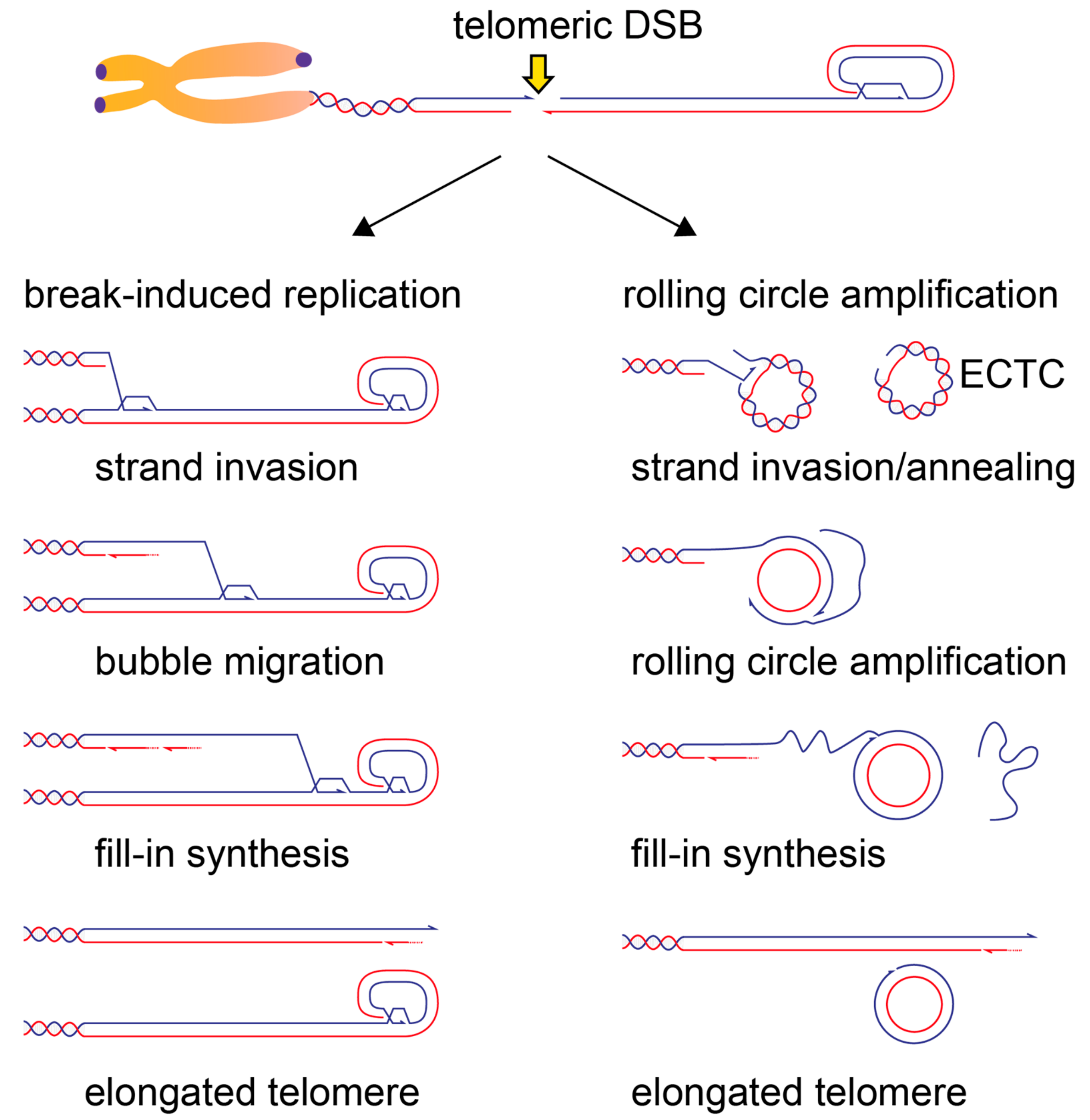
8. Telomere Elongation by Homologous Recombination
9. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, I.; de Lange, T. Shelterin. Curr. Biol. 2016, 26, R397–R399. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cech, T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 2001, 292, 1171–1175. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Daniels, J.P.; Takai, H.; de Lange, T. Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell 2006, 126, 63–77. [Google Scholar] [CrossRef]
- Wu, L.; Multani, A.S.; He, H.; Cosme-Blanco, W.; Deng, Y.; Deng, J.M.; Bachilo, O.; Pathak, S.; Tahara, H.; Bailey, S.M.; et al. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 2006, 126, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; de Lange, T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 2014, 6, a016576. [Google Scholar] [CrossRef]
- Van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 1999, 283, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Karlseder, J.; Holtgreve-Grez, H.; Jauch, A.; de Lange, T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 2002, 12, 1635–1644. [Google Scholar] [CrossRef]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell. Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Sfeir, A.J.; Shay, J.W.; Wright, W.E.; de Lange, T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005, 24, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.; Lukas, J.; Jackson, S. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell. Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Arnoult, N.; Saintome, C.; Ourliac-Garnier, I.; Riou, J.F.; Londono-Vallejo, A. Human POT1 is required for efficient telomere C-rich strand replication in the absence of WRN. Genes Dev. 2009, 23, 2915–2924. [Google Scholar] [CrossRef]
- Pinzaru, A.M.; Hom, R.A.; Beal, A.; Phillips, A.F.; Ni, E.; Cardozo, T.; Nair, N.; Choi, J.; Wuttke, D.S.; Sfeir, A.; et al. Telomere Replication Stress Induced by POT1 Inactivation Accelerates Tumorigenesis. Cell Rep. 2016, 15, 2170–2184. [Google Scholar] [CrossRef]
- Gong, Y.; de Lange, T. A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell. 2010, 40, 377–387. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Palm, W.; Else, T.; Daniels, J.P.; Takai, K.K.; Ye, J.Z.; Keegan, C.E.; de Lange, T.; Hammer, G.D. Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat. Struct. Mol. Biol. 2007, 14, 754–761. [Google Scholar] [CrossRef]
- Takai, K.K.; Kibe, T.; Donigian, J.R.; Frescas, D.; de Lange, T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol. Cell. 2011, 44, 647–659. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Stansel, R.M.; de Lange, T.; Griffith, J.D. T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef]
- Amiard, S.; Doudeau, M.; Pinte, S.; Poulet, A.; Lenain, C.; Faivre-Moskalenko, C.; Angelov, D.; Hug, N.; Vindigni, A.; Bouvet, P.; et al. A topological mechanism for TRF2-enhanced strand invasion. Nat. Struct. Mol. Biol. 2007, 14, 147–154. [Google Scholar] [CrossRef]
- Murti, K.G.; Prescott, D.M. Telomeres of polytene chromosomes in a ciliated protozoan terminate in duplex DNA loops. Proc. Natl. Acad. Sci. USA 1999, 96, 14436–14439. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Cross, G.A.; de Lange, T.; Griffith, J.D. t-loops at trypanosome telomeres. EMBO J. 2001, 20, 579–588. [Google Scholar] [CrossRef]
- Cesare, A.J.; Quinney, N.; Willcox, S.; Subramanian, D.; Griffith, J.D. Telomere looping in P. sativum (common garden pea). Plant J. 2003, 36, 271–279. [Google Scholar] [CrossRef]
- Nikitina, T.; Woodcock, C.L. Closed chromatin loops at the ends of chromosomes. J. Cell Biol. 2004, 166, 161–165. [Google Scholar] [CrossRef]
- Cesare, A.J.; Groff-Vindman, C.; Compton, S.A.; McEachern, M.J.; Griffith, J.D. Telomere loops and homologous recombination-dependent telomeric circles in a Kluyveromyces lactis telomere mutant strain. Mol. Cell. Biol. 2008, 28, 20–29. [Google Scholar] [CrossRef]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef]
- Van Ly, D.; Low, R.R.J.; Frolich, S.; Bartolec, T.K.; Kafer, G.R.; Pickett, H.A.; Gaus, K.; Cesare, A.J. Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell. 2018, 71, 510–525. [Google Scholar] [CrossRef]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, J.R., 3rd; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef]
- Ribes-Zamora, A.; Indiviglio, S.M.; Mihalek, I.; Williams, C.L.; Bertuch, A.A. TRF2 interaction with Ku heterotetramerization interface gives insight into c-NHEJ prevention at human telomeres. Cell. Rep. 2013, 5, 194–206. [Google Scholar] [CrossRef]
- Takai, K.K.; Hooper, S.; Blackwood, S.; Gandhi, R.; de Lange, T. In vivo stoichiometry of shelterin components. J. Biol. Chem. 2010, 285, 1457–1467. [Google Scholar] [CrossRef]
- Lyamichev, V.I.; Mirkin, S.M.; Danilevskaya, O.N.; Voloshin, O.N.; Balatskaya, S.V.; Dobrynin, V.N.; Filippov, S.A.; Frank-Kamenetskii, M.D. An unusual DNA structure detected in a telomeric sequence under superhelical stress and at low pH. Nature 1989, 339, 634–637. [Google Scholar] [CrossRef]
- Veselkov, A.G.; Malkov, V.A.; Frank-Kamenetskll, M.D.; Dobrynin, V.N. Triplex model of chromosome ends. Nature 1993, 364, 496. [Google Scholar] [CrossRef]
- Paeschke, K.; Simonsson, T.; Postberg, J.; Rhodes, D.; Lipps, H.J. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat. Struct. Mol. Biol. 2005, 12, 847–854. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell. Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef]
- Doksani, Y.; de Lange, T. Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell Rep. 2016, 17, 1646–1656. [Google Scholar] [CrossRef]
- Mao, P.; Liu, J.; Zhang, Z.; Zhang, H.; Liu, H.; Gao, S.; Rong, Y.S.; Zhao, Y. Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nat. Commun. 2016, 7, 12154. [Google Scholar] [CrossRef]
- Lam, Y.C.; Akhter, S.; Gu, P.; Ye, J.; Poulet, A.; Giraud-Panis, M.J.; Bailey, S.M.; Gilson, E.; Legerski, R.J.; Chang, S. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 2010, 29, 2230–2241. [Google Scholar] [CrossRef]
- Wu, P.; van Overbeek, M.; Rooney, S.; de Lange, T. Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol. Cell. 2010, 39, 606–617. [Google Scholar] [CrossRef]
- Wu, P.; Takai, H.; de Lange, T. Telomeric 3’ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 2012, 150, 39–52. [Google Scholar] [CrossRef]
- Bae, N.S.; Baumann, P. A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol. Cell. 2007, 26, 323–334. [Google Scholar] [CrossRef]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Benarroch-Popivker, D.; Pisano, S.; Mendez-Bermudez, A.; Lototska, L.; Kaur, P.; Bauwens, S.; Djerbi, N.; Latrick, C.M.; Fraisier, V.; Pei, B.; et al. TRF2-Mediated Control of Telomere DNA Topology as a Mechanism for Chromosome-End Protection. Mol. Cell. 2016, 61, 274–286. [Google Scholar] [CrossRef]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 2017, 549, 548–552. [Google Scholar] [CrossRef]
- Sarthy, J.; Bae, N.S.; Scrafford, J.; Baumann, P. Human RAP1 inhibits non-homologous end joining at telomeres. EMBO J. 2009, 28, 3390–3399. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair (Amst.) 2014, 17, 81–97. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Fishman-Lobell, J.; Rudin, N.; Haber, J.E. Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell. Biol. 1992, 12, 1292–1303. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef]
- Rai, R.; Zheng, H.; He, H.; Luo, Y.; Multani, A.; Carpenter, P.B.; Chang, S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010, 29, 2598–2610. [Google Scholar] [CrossRef]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Schmutz, I.; Timashev, L.; Xie, W.; Patel, D.J.; de Lange, T. TRF2 binds branched DNA to safeguard telomere integrity. Nat. Struct. Mol. Biol. 2017, 24, 734–742. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar] [CrossRef]
- Maser, R.S.; Wong, K.K.; Sahin, E.; Xia, H.; Naylor, M.; Hedberg, H.M.; Artandi, S.E.; DePinho, R.A. DNA-dependent protein kinase catalytic subunit is not required for dysfunctional telomere fusion and checkpoint response in the telomerase-deficient mouse. Mol. Cell. Biol 2007, 27, 2253–2265. [Google Scholar] [CrossRef]
- Letsolo, B.T.; Rowson, J.; Baird, D.M. Fusion of short telomeres in human cells is characterized by extensive deletion and microhomology, and can result in complex rearrangements. Nucleic Acids Res. 2010, 38, 1841–1852. [Google Scholar] [CrossRef]
- Jones, R.E.; Oh, S.; Grimstead, J.W.; Zimbric, J.; Roger, L.; Heppel, N.H.; Ashelford, K.E.; Liddiard, K.; Hendrickson, E.A.; Baird, D.M. Escape from telomere-driven crisis is DNA ligase III dependent. Cell Rep. 2014, 8, 1063–1076. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst.) 2016, 44, 68–75. [Google Scholar] [CrossRef]
- West, S.C.; Blanco, M.G.; Chan, Y.W.; Matos, J.; Sarbajna, S.; Wyatt, H.D. Resolution of Recombination Intermediates: Mechanisms and Regulation. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 103–109. [Google Scholar] [CrossRef]
- Bailey, S.M.; Goodwin, E.H.; Cornforth, M.N. Strand-specific fluorescence in situ hybridization: The CO-FISH family. Cytogenet. Genome Res. 2004, 107, 14–17. [Google Scholar] [CrossRef]
- Chaganti, R.; Schonberg, S.; German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 71, 4508–4512. [Google Scholar] [CrossRef]
- Van Wietmarschen, N.; Lansdorp, P.M. Bromodeoxyuridine does not contribute to sister chromatid exchange events in normal or Bloom syndrome cells. Nucleic Acids Res. 2016, 44, 6787–6793. [Google Scholar] [CrossRef]
- Kato, H.; Stich, H.F. Sister chromatid exchanges in ageing and repair-deficient human fibroblasts. Nature 1976, 260, 447–448. [Google Scholar] [CrossRef]
- Allen, J.W.; Latt, S.A. Analysis of sister chromatid exchange formation in vivo in mouse spermatogonia as a new test system for environmental mutagens. Nature 1976, 260, 449–451. [Google Scholar] [CrossRef]
- Vogel, W.; Bauknecht, T. Differential chromatid staining by in vivo treatment as a mutagenicity test system. Nature 1976, 260, 448–449. [Google Scholar] [CrossRef]
- Laud, P.R.; Multani, A.S.; Bailey, S.M.; Wu, L.; Ma, J.; Kingsley, C.; Lebel, M.; Pathak, S.; DePinho, R.A.; Chang, S. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005, 19, 2560–2570. [Google Scholar] [CrossRef]
- Celli, G.B.; Denchi, E.L.; de Lange, T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 2006, 8, 885–890. [Google Scholar] [CrossRef]
- Palm, W.; Hockemeyer, D.; Kibe, T.; de Lange, T. Functional dissection of human and mouse POT1 proteins. Mol. Cell. Biol 2009, 29, 471–482. [Google Scholar] [CrossRef]
- Sfeir, A.; Kabir, S.; van Overbeek, M.; Celli, G.B.; de Lange, T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 2010, 327, 1657–1661. [Google Scholar] [CrossRef]
- Kabir, S.; Hockemeyer, D.; de Lange, T. TALEN gene knockouts reveal no requirement for the conserved human shelterin protein Rap1 in telomere protection and length regulation. Cell Rep. 2014, 9, 1273–1280. [Google Scholar] [CrossRef]
- Rai, R.; Chen, Y.; Lei, M.; Chang, S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun. 2016, 7, 10881. [Google Scholar] [CrossRef]
- Cornforth, M.N.; Eberle, R.L. Termini of human chromosomes display elevated rates of mitotic recombination. Mutagenesis 2001, 16, 85–89. [Google Scholar] [CrossRef]
- Bailey, S.M.; Brenneman, M.A.; Goodwin, E.H. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004, 32, 3743–3751. [Google Scholar] [CrossRef]
- Rudd, M.K.; Friedman, C.; Parghi, S.S.; Linardopoulou, E.V.; Hsu, L.; Trask, B.J. Elevated rates of sister chromatid exchange at chromosome ends. PLoS Genet. 2007, 3, e32. [Google Scholar] [CrossRef]
- Mozlin, A.M.; Fung, C.W.; Symington, L.S. Role of the Saccharomyces cerevisiae Rad51 paralogs in sister chromatid recombination. Genetics 2008, 178, 113–126. [Google Scholar] [CrossRef]
- Adar, S.; Izhar, L.; Hendel, A.; Geacintov, N.; Livneh, Z. Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res. 2009, 37, 5737–5748. [Google Scholar] [CrossRef]
- Ma, W.; Westmoreland, J.W.; Resnick, M.A. Homologous recombination rescues ssDNA gaps generated by nucleotide excision repair and reduced translesion DNA synthesis in yeast G2 cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2895–2904. [Google Scholar] [CrossRef]
- Vriend, L.E.; Krawczyk, P.M. Nick-initiated homologous recombination: Protecting the genome, one strand at a time. DNA Repair (Amst.) 2017, 50, 1–13. [Google Scholar] [CrossRef]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef]
- Wang, R.C.; Smogorzewska, A.; de Lange, T. Homologous Recombination Generates T-Loop-Sized Deletions at Human Telomeres. Cell 2004, 119, 355–368. [Google Scholar] [CrossRef]
- Greenberg, R.A.; Chin, L.; Femino, A.; Lee, K.H.; Gottlieb, G.J.; Singer, R.H.; Greider, C.W.; DePinho, R.A. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 1999, 97, 515–525. [Google Scholar] [CrossRef]
- Qi, L.; Strong, M.A.; Karim, B.O.; Armanios, M.; Huso, D.L.; Greider, C.W. Short telomeres and ataxia-telangiectasia mutated deficiency cooperatively increase telomere dysfunction and suppress tumorigenesis. Cancer Res. 2003, 63, 8188–8196. [Google Scholar]
- Feldser, D.M.; Greider, C.W. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007, 11, 461–469. [Google Scholar] [CrossRef]
- Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W., 2nd; Greider, C.W.; DePinho, R.A. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell. Biol 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Huffman, K.E.; Levene, S.D.; Tesmer, V.M.; Shay, J.W.; Wright, W.E. Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J. Biol. Chem. 2000, 275, 19719–19722. [Google Scholar] [CrossRef]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Bar-Or, D.; Thomas, G.W.; Rael, L.T.; Lau, E.P.; Winkler, J.V. Asp-Ala-His-Lys (DAHK) inhibits copper-induced oxidative DNA double strand breaks and telomere shortening. Biochem. Biophys. Res. Commun. 2001, 282, 356–360. [Google Scholar] [CrossRef]
- Oikawa, S.; Tada-Oikawa, S.; Kawanishi, S. Site-Specific DNA Damage at the GGG Sequence by UVA Involves Acceleration of Telomere Shortening. Biochemistry 2001, 40, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Saretzki, G.; Docke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Sitte, N.; Saretzki, G.; von Zglinicki, T. Accelerated telomere shortening in fibroblasts after extended periods of confluency. Free Radic. Biol. Med. 1998, 24, 885–893. [Google Scholar] [CrossRef]
- Munro, J.; Steeghs, K.; Morrison, V.; Ireland, H.; Parkinson, E.K. Human fibroblast replicative senescence can occur in the absence of extensive cell division and short telomeres. Oncogene 2001, 20, 3541–3552. [Google Scholar] [CrossRef]
- Martinez, P.; Thanasoula, M.; Munoz, P.; Liao, C.; Tejera, A.; McNees, C.; Flores, J.M.; Fernandez-Capetillo, O.; Tarsounas, M.; Blasco, M.A. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009, 23, 2060–2075. [Google Scholar] [CrossRef]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef]
- Miller, K.M.; Rog, O.; Cooper, J.P. Semi-conservative DNA replication through telomeres requires Taz1. Nature 2006, 440, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Iwano, T.; Tachibana, M.; Reth, M.; Shinkai, Y. Importance of TRF1 for functional telomere structure. J. Biol. Chem. 2004, 279, 1442–1448. [Google Scholar] [CrossRef]
- Vannier, J.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.; Ding, H.; Boulton, S. RTEL1 Dismantles T Loops and Counteracts Telomeric G4-DNA to Maintain Telomere Integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Kibe, T.; Kabir, S.; de Lange, T. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 2014, 28, 2477–2491. [Google Scholar] [CrossRef]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 2004, 306, 1951–1953. [Google Scholar] [CrossRef]
- Sarek, G.; Vannier, J.B.; Panier, S.; Petrini, J.H.J.; Boulton, S.J. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol. Cell. 2015, 57, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Opresko, P.L.; von Kobbe, C.; Laine, J.P.; Harrigan, J.; Hickson, I.D.; Bohr, V.A. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J. Biol. Chem. 2002, 277, 41110–41119. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Orren, D.K. TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNA. Oncogene 2004, 23, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Diotti, R.; Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2011, 2, 119–135. [Google Scholar] [CrossRef]
- Hemann, M.T.; Strong, M.A.; Hao, L.-Y.; Greider, C.W. The Shortest Telomere, Not Average Telomere Length, Is Critical for Cell Viability and Chromosome Stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Sabatier, L.; Ricoul, M.; Pottier, G.; Murnane, J.P. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005, 3, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Sfeir, A.; Gryaznov, S.M.; Shay, J.W.; Wright, W.E. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol. Biol. Cell 2004, 15, 3709–3718. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.P.; Zhang, N.; Noh, J.; Mender, I.; Tedone, E.; Huang, E.; Wright, W.E.; Danuser, G.; Shay, J.W. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat. Commun. 2017, 8, 1356. [Google Scholar] [CrossRef]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef]
- Londono-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol 2017, 37. [Google Scholar] [CrossRef]
- Gocha, A.R.; Harris, J.; Groden, J. Alternative mechanisms of telomere lengthening: Permissive mutations, DNA repair proteins and tumorigenic progression. Mutat. Res. 2013, 743–744, 142–150. [Google Scholar] [CrossRef]
- Bechter, O.E.; Shay, J.W.; Wright, W.E. The frequency of homologous recombination in human ALT cells. Cell Cycle 2004, 3, 547–549. [Google Scholar] [CrossRef]
- Lovejoy, C.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.; Sung, P.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, 1–16. [Google Scholar] [CrossRef]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef]
- Nabetani, A.; Ishikawa, F. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol. Cell. Biol. 2009, 29, 703–713. [Google Scholar] [CrossRef]
- Fasching, C.L.; Neumann, A.A.; Muntoni, A.; Yeager, T.R.; Reddel, R.R. DNA damage induces alternative lengthening of telomeres (ALT) associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res. 2007, 67, 7072–7077. [Google Scholar] [CrossRef]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef]
- Henle, E.S.; Han, Z.X.; Tang, N.; Rai, P.; Luo, Y.Z.; Linn, S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J. Biol. Chem. 1999, 274, 962–971. [Google Scholar] [CrossRef]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT Cells Is Characterized by Free Telomeric Circles and Heterogeneous t-Loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Tomaska, L.; McEachern, M.J.; Nosek, J. Alternatives to telomerase: Keeping linear chromosomes via telomeric circles. FEBS Lett. 2004, 567, 142–146. [Google Scholar] [CrossRef]
- Fouche, N.; Cesare, A.; Willcox, S.; Ozgur, S.; Compton, S.; Griffith, J. The basic domain of TRF2 directs binding to DNA junctions irrespective of the presence of TTAGGG repeats. J. Biol. Chem. 2006, 281, 37486–37495. [Google Scholar] [CrossRef]
- Poulet, A.; Buisson, R.; Faivre-Moskalenko, C.; Koelblen, M.; Amiard, S.; Montel, F.; Cuesta-Lopez, S.; Bornet, O.; Guerlesquin, F.; Godet, T.; et al. TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J. 2009, 28, 641–651. [Google Scholar] [CrossRef]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef]
- Wang, Y.; Ghosh, G.; Hendrickson, E.A. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12430–12435. [Google Scholar] [CrossRef]
- Gu, P.; Min, J.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012, 31, 2309–2321. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Dheekollu, J.; Broccoli, D.; Dutta, A.; Lieberman, P.M. The origin recognition complex localizes to telomere repeats and prevents telomere-circle formation. Curr. Biol. 2007, 17, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Li, J.S.; Miralles Fuste, J.; Simavorian, T.; Bartocci, C.; Tsai, J.; Karlseder, J.; Lazzerini Denchi, E. TZAP: A telomere-associated protein involved in telomere length control. Science 2017, 355, 638–641. [Google Scholar] [CrossRef]
- Tomaska, L.; Nosek, J.; Kramara, J.; Griffith, J.D. Telomeric circles: Universal players in telomere maintenance? Nat. Struct. Mol. Biol. 2009, 16, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Regev, A.; Cohen, S.; Cohen, E.; Bar-Am, I.; Lavi, S. Telomeric repeats on small polydisperse circular DNA (spcDNA) and genomic instability. Oncogene 1998, 17, 3455–3461. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Shengzhao, G.; Li, X.; Liu, H.; Zhao, Y. Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet. 2019, 15, e1007925. [Google Scholar] [CrossRef]
- Rivera, T.; Haggblom, C.; Cosconati, S.; Karlseder, J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2016, 24, 30–39. [Google Scholar] [CrossRef]
- Poole, L.A.; Zhao, R.; Glick, G.G.; Lovejoy, C.A.; Eischen, C.M.; Cortez, D. SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 14864–14869. [Google Scholar] [CrossRef] [PubMed]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef]
- Wilson, M.A.; Kwon, Y.; Xu, Y.; Chung, W.H.; Chi, P.; Niu, H.; Mayle, R.; Chen, X.; Malkova, A.; Sung, P.; et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 2013, 502, 393–396. [Google Scholar] [CrossRef]
- Nosek, J.; Rycovska, A.; Makhov, A.M.; Griffith, J.D.; Tomaska, L. Amplification of telomeric arrays via rolling-circle mechanism. J. Biol. Chem. 2005, 280, 10840–10845. [Google Scholar] [CrossRef]
- Muntoni, A.; Neumann, A.A.; Hills, M.; Reddel, R.R. Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum. Mol. Genet. 2009, 18, 1017–1027. [Google Scholar] [CrossRef]





© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doksani, Y. The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes 2019, 10, 318. https://doi.org/10.3390/genes10040318
Doksani Y. The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes. 2019; 10(4):318. https://doi.org/10.3390/genes10040318
Chicago/Turabian StyleDoksani, Ylli. 2019. "The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function" Genes 10, no. 4: 318. https://doi.org/10.3390/genes10040318
APA StyleDoksani, Y. (2019). The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes, 10(4), 318. https://doi.org/10.3390/genes10040318