Viral Metagenomics on Cerebrospinal Fluid

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
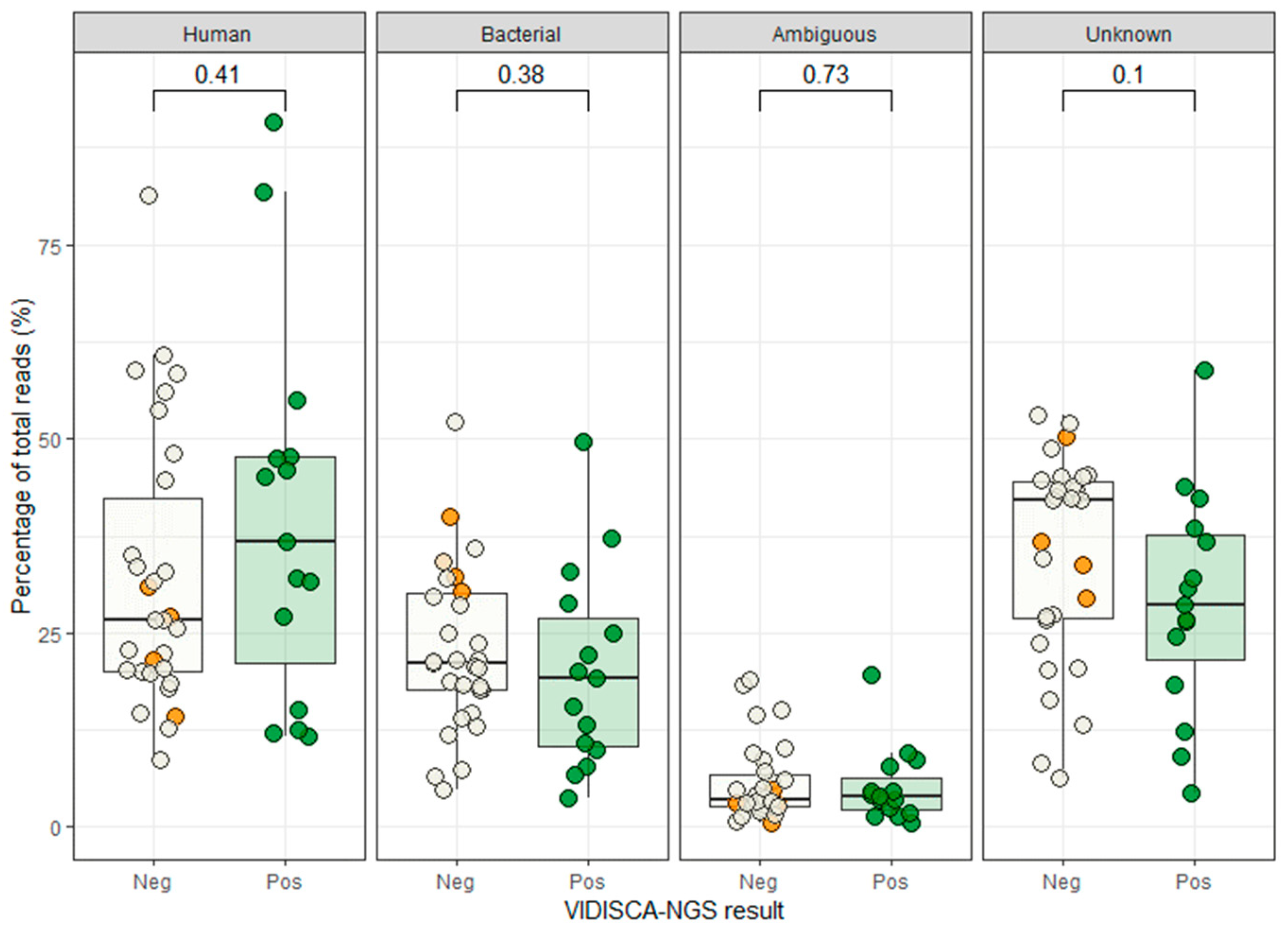
3. Results
3.1. Sample Description and qPCR Results
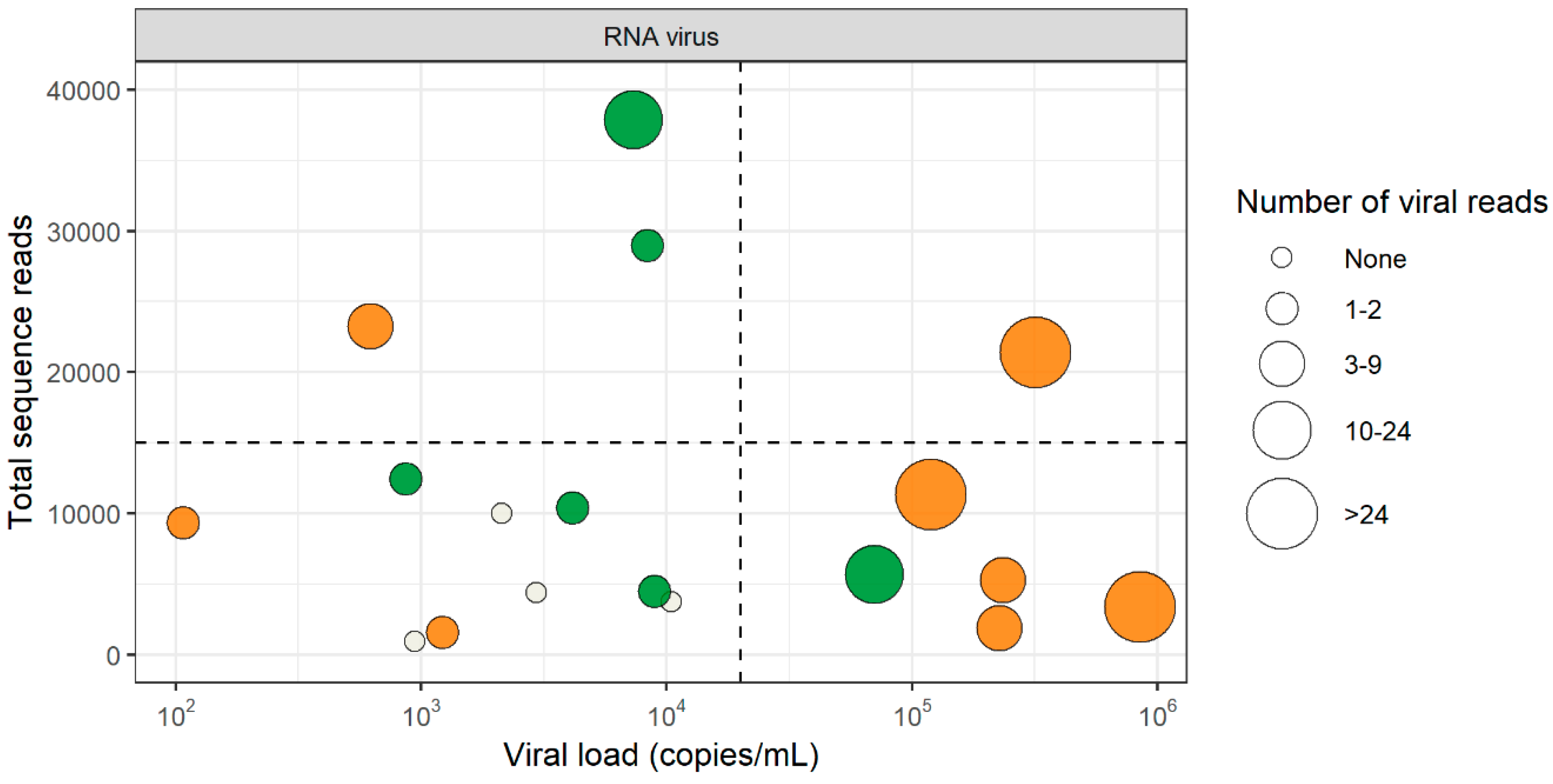
3.2. RNA Virus Detection by VIDISCA-NGS
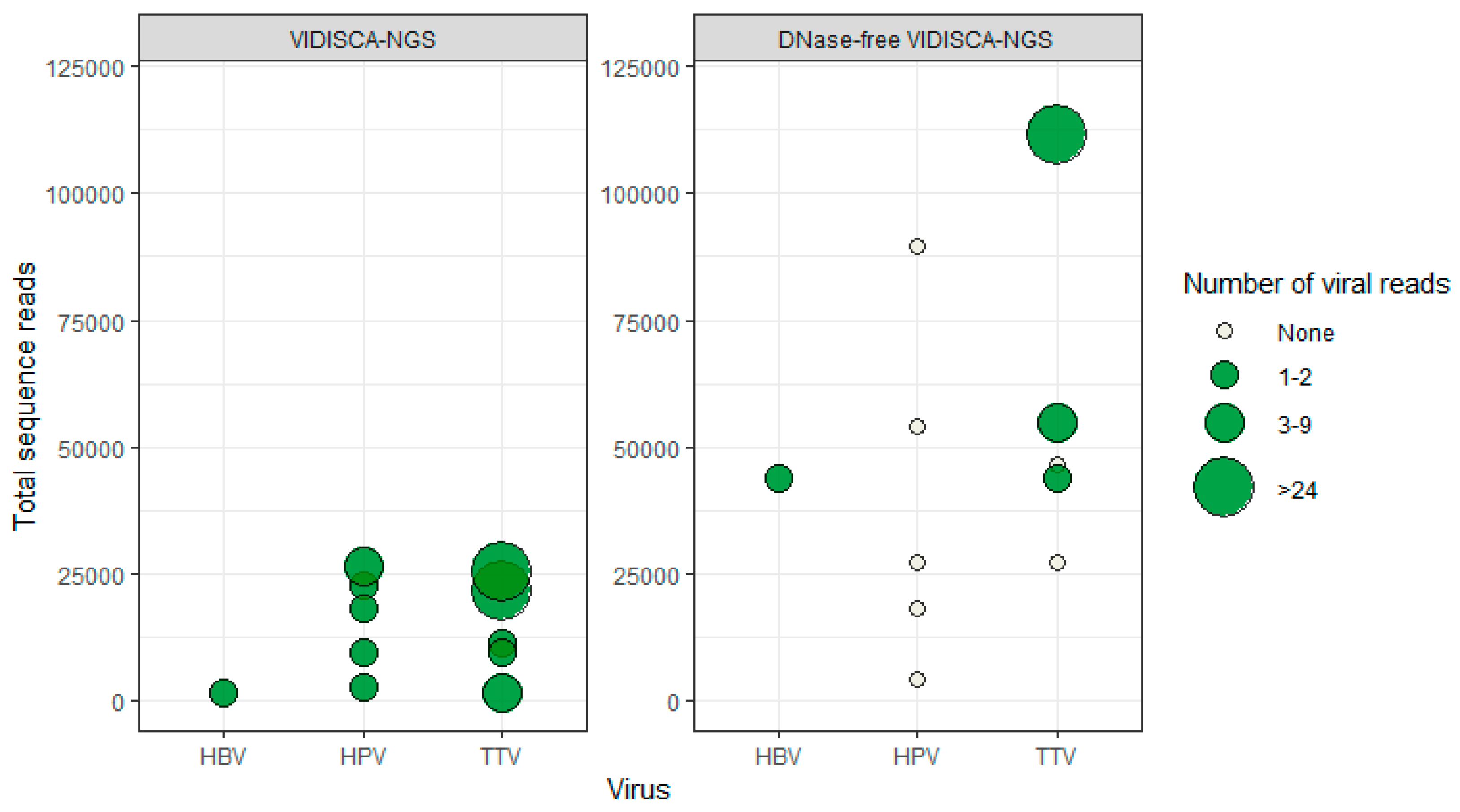
3.3. DNA Virus Detection by VIDISCA-NGS
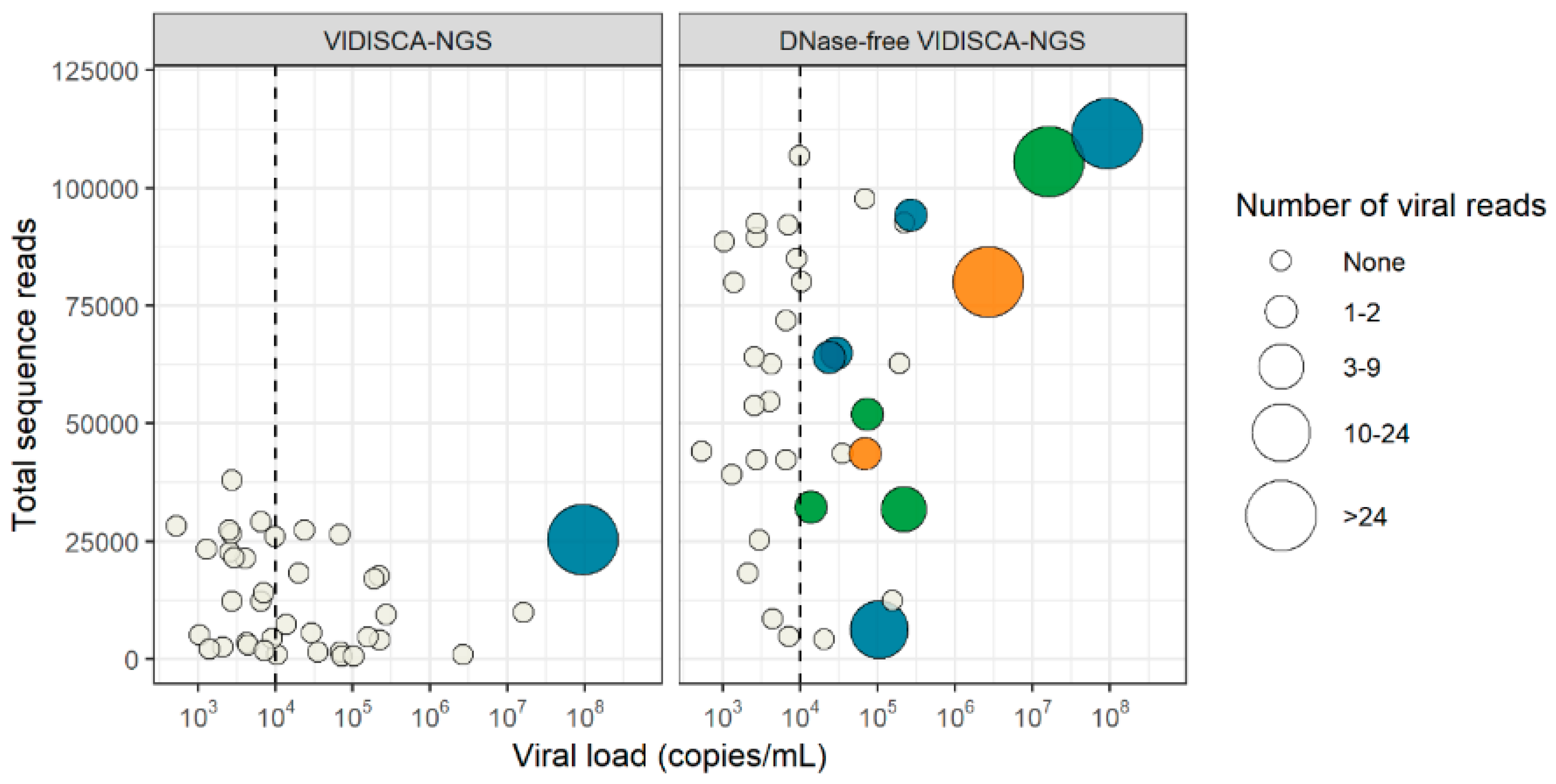
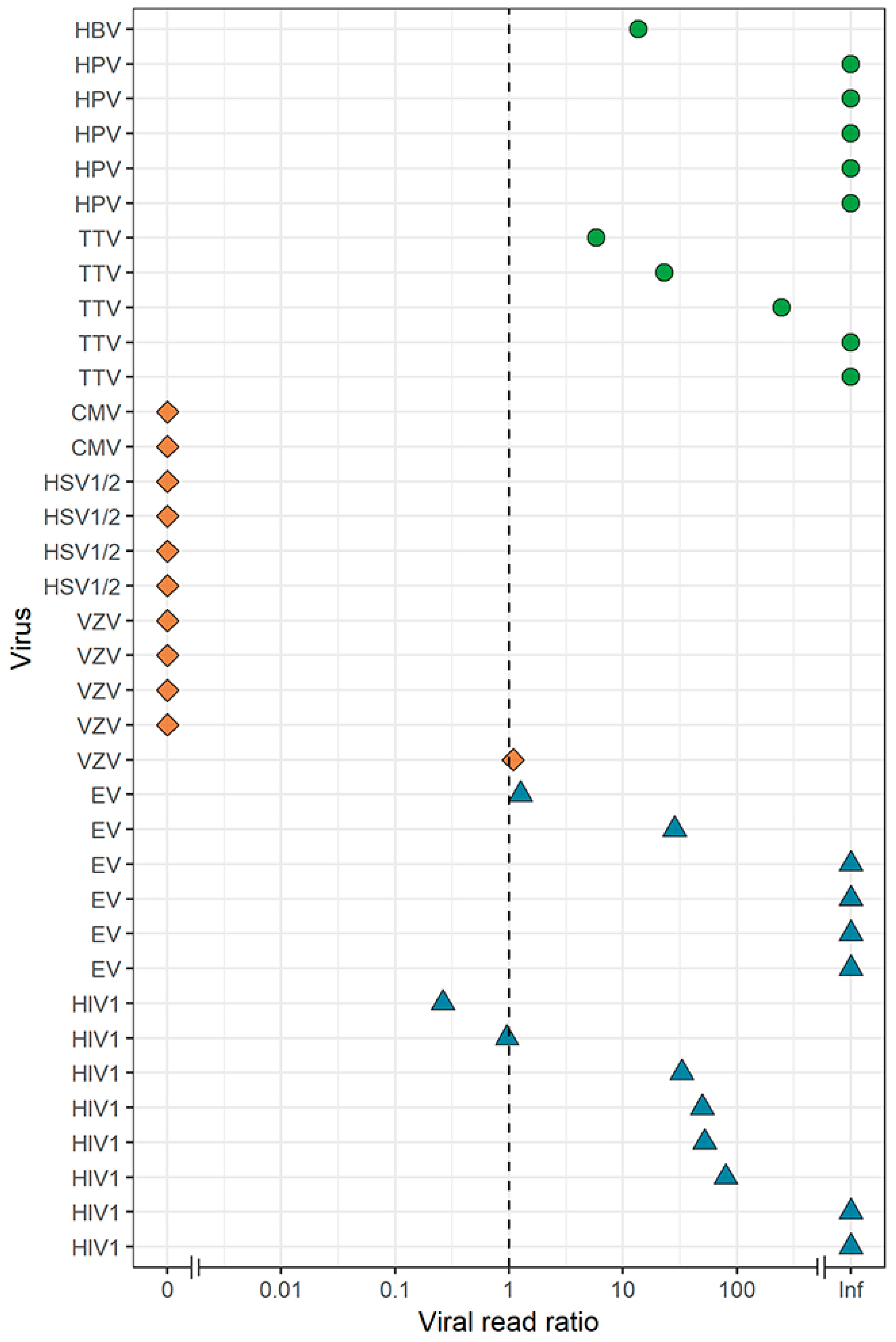
3.4. Virus Detection by DNase-Free VIDISCA-NGS
3.5. Effect of a DNase Treatment on Virus Detection by VIDISCA-NGS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brouwer, M.C.; Thwaites, G.E.; Tunkel, A.R.; van de Beek, D. Dilemmas in the diagnosis of acute community-acquired bacterial meningitis. Lancet 2012, 380, 1684–1692. [Google Scholar] [CrossRef]
- Khatib, U.; van de Beek, D.; Lees, J.A.; Brouwer, M.C. Adults with suspected central nervous system infection: A prospective study of diagnostic accuracy. J. Infect. 2017, 74, 1–9. [Google Scholar] [CrossRef]
- Solomon, T.; Hart, I.J.; Beeching, N.J. Viral encephalitis: A clinician’s guide. Pract. Neurol. 2007, 7, 288–305. [Google Scholar] [CrossRef]
- Granerod, J.; Tam, C.C.; Crowcroft, N.S.; Davies, N.W.S.; Borchert, M.; Thomas, S.L. Challenge of the unknown: A systematic review of acute encephalitis in non-outbreak situations. Neurology 2010, 75, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Bharucha, T.; Breuer, J. Encephalitis diagnosis using metagenomics: Application of next generation sequencing for undiagnosed cases. J. Infect. 2018, 76, 225–240. [Google Scholar] [CrossRef]
- Whitley, R.J.; Gnann, J.W. Viral encephalitis: Familiar infections and emerging pathogens. Lancet 2002, 359, 507–514. [Google Scholar] [CrossRef]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A New Arenavirus in a Cluster of Fatal Transplant-Associated Diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef]
- Hoffmann, B.; Tappe, D.; Höper, D.; Herden, C.; Boldt, A.; Mawrin, C.; Niederstraßer, O.; Müller, T.; Jenckel, M.; van der Grinten, E.; et al. A Variegated Squirrel Bornavirus Associated with Fatal Human Encephalitis. N. Engl. J. Med. 2015, 373, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Edridge, A.W.D.; Deijs, M.; Namazzi, R.; Cristella, C.; Jebbink, M.F.; Maurer, I.; Kootstra, N.A.; Buluma, L.R.; van Woensel, J.B.M.; de Jong, M.D.; et al. Novel Orthobunyavirus Identified in the Cerebrospinal Fluid of a Ugandan Child With Severe Encephalopathy. Clin. Infect. Dis. 2019, 68. [Google Scholar] [CrossRef]
- Quan, P.L.; Wagner, T.A.; Briese, T.; Torgerson, T.R.; Hornig, M.; Tashmukhamedova, A.; Firth, C.; Palacios, G.; Baisre-de-Leon, A.; Paddock, C.D.; et al. Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerg. Infect. Dis. 2010, 16, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Conceição Neto, N.; Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; et al. NetoVIR: a reproducible protocol for virome analysis. Protoc. Exch. 2016. [Google Scholar] [CrossRef]
- Wylezich, C.; Papa, A.; Beer, M.; Höper, D. A Versatile Sample Processing Workflow for Metagenomic Pathogen Detection. Sci. Rep. 2018, 8, 13108. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.M.; Wolthers, K.C.; Wertheim-van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Williams, C.V.; Gadi, S.R.; Jebbink, M.F.; Oude Munnink, B.B.; Jazaeri Farsani, S.M.; Cullen, J.M.; van der Hoek, L. Persistent viremia by a novel parvovirus in a slow loris (Nycticebus coucang) with diffuse histiocytic sarcoma. Front. Microbiol. 2014, 5, 655. [Google Scholar] [CrossRef]
- Canuti, M.; Williams, C.V.; Sagan, S.M.; Oude Munnink, B.B.; Gadi, S.; Verhoeven, J.T.P.; Kellam, P.; Cotten, M.; Lang, A.S.; Junge, R.E.; et al. Virus discovery reveals frequent infection by diverse novel members of the Flaviviridae in wild lemurs. Arch. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.; Oude Munnink, B.B.; Deijs, M.; Canuti, M.; Koekkoek, S.M.; Molenkamp, R.; Bakker, M.; Jurriaans, S.; van Schaik, B.D.C.; Luyf, A.C.; et al. Performance of VIDISCA-454 in Feces-Suspensions and Serum. Viruses 2012, 4, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.; Deijs, M.; Canuti, M.; van Schaik, B.D.C.; Faria, N.R.; van de Garde, M.D.B.; Jachimowski, L.C.M.; Jebbink, M.F.; Jakobs, M.; Luyf, A.C.M.; et al. A sensitive assay for virus discovery in respiratory clinical samples. PLoS ONE 2011, 6, e16118. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.; de Vries, M.; van Steenbeek, F.G.; Favier, R.P.; Deijs, M.; Brinkhof, B.; Rothuizen, J.; van der Hoek, L.; Penning, L.C. Sequence-independent VIDISCA-454 technique to discover new viruses in canine livers. J. Virol. Methods 2012, 185, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.C.; Jim, K.K.; Benschop, K.S.; Wolthers, K.C.; van der Ende, A.; de Jong, M.D.; van de Beek, D. No evidence of viral coinfection in cerebrospinal fluid from patients with community-acquired bacterial meningitis. J. Infect. Dis. 2013, 208, 182–184. [Google Scholar] [CrossRef]
- Boom, R.; Sol, C.J.A.; Salimans, M.M.M.; Jansen, C.L.; Wertheim-van Dillen, P.M.E.; van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar]
- Endoh, D.; Mizutani, T.; Kirisawa, R.; Maki, Y.; Saito, H.; Kon, Y.; Morikawa, S.; Hayashi, M. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 2005, 33, 1–11. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Kinsella, C.M.; Deijs, M.; van der Hoek, L. Enhanced bioinformatic profiling of VIDISCA libraries for virus detection and discovery. Virus Res. 2018, 263, 21–26. [Google Scholar] [CrossRef]
- Flygare, S.; Simmon, K.; Miller, C.; Qiao, Y.; Kennedy, B.; Di Sera, T.; Graf, E.H.; Tardif, K.D.; Kapusta, A.; Rynearson, S.; et al. Taxonomer: An interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome Biol. 2016, 17, 111. [Google Scholar] [CrossRef]
- Bukowska-Ośko, I.; Perlejewski, K.; Nakamura, S.; Motooka, D.; Stokowy, T.; Kosińska, J.; Popiel, M.; Płoski, R.; Horban, A.; Lipowski, D.; et al. Sensitivity of next-generation sequencing metagenomic analysis for detection of RNA and DNA viruses in cerebrospinal fluid: The confounding effect of background contamination. In Respiratory Treatment and Prevention; Advances in Experimental Medicine and Biology; Springer: Cham, Germany, 2017; Volume 944, pp. 53–62. ISBN 978-3-319-44488-8. [Google Scholar] [CrossRef]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G.; Professional Practice Committee and Committee on Laboratory Practices of the American Society for Microbiology and Microbiology Resource Committee of the College of American Pathologists. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory Validation of a Clinical Metagenomic Sequencing Assay for Pathogen Detection in Cerebrospinal Fluid. bioRxiv 2018, 330381. [Google Scholar] [CrossRef]
- De Groof, A.; Deijs, M.; Guelen, L.; van Grinsven, L.; van Os-Galdos, L.; Vogels, W.; Derks, C.; Cruijsen, T.; Geurts, V.; Vrijenhoek, M.; et al. Atypical Porcine Pestivirus: A Possible Cause of Congenital Tremor Type A-II in Newborn Piglets. Viruses 2016, 8, 271. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Cotten, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; van Hemert, F.J.; Phan, M.V.T.; Bakker, M.; Jazaeri Farsani, S.M.; Kellam, P.; et al. A Novel Astrovirus-Like RNA Virus Detected in Human Stool. Virus Evol. 2016, 2, vew005. [Google Scholar] [CrossRef]
- de Groof, A.; Guelen, L.; Deijs, M.; van der Wal, Y.; Miyata, M.; Ng, K.S.; van Grinsven, L.; Simmelink, B.; Biermann, Y.; Grisez, L.; et al. A Novel Virus Causes Scale Drop Disease in Lates calcarifer. PLoS Pathog. 2015, 11, e1005074. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Canuti, M.; Deijs, M.; de Vries, M.; Jebbink, M.F.; Rebers, S.; Molenkamp, R.; van Hemert, F.J.; Chung, K.; Cotten, M.; et al. Unexplained diarrhoea in HIV-1 infected individuals. BMC Infect. Dis. 2014, 14, 22. [Google Scholar] [CrossRef]
- Jazaeri Farsani, S.M.; Jebbink, M.F.; Deijs, M.; Canuti, M.; van Dort, K.A.; Bakker, M.; Grady, B.P.; Prins, M.; van Hemert, F.J.; Kootstra, N.A.; et al. Identification of a new genotype of Torque Teno Mini virus. Virol. J. 2013, 10, 323. [Google Scholar] [CrossRef]
- Boom, R.; Sol, C.J.A.; Schuurman, T.; van Breda, A.; Weel, J.F.L.; Beld, M.; Ten Berge, I.J.M.; Wertheim-van Dillen, P.M.E.; de Jong, M.D. Human cytomegalovirus DNA in plasma and serum specimens of renal transplant recipients is highly fragmented. J. Clin. Microbiol. 2002, 40, 4105–4113. [Google Scholar] [CrossRef]
- Stokowy, T.; Bukowska-Ośko, I.; Nakamura, S.; Pollak, A.; Lechowicz, U.; Popiel, M.; Caraballo Cortés, K.; Radkowski, M.; Lipowski, D.; Motooka, D.; et al. Next-generation sequencing (NGS) in the identification of encephalitis-causing viruses: Unexpected detection of human herpesvirus 1 while searching for RNA pathogens. J. Virol. Methods 2015, 226, 1–6. [Google Scholar]
- Huang, E.S.; Chen, S.T.; Pagano, J.S. Human cytomegalovirus. I. Purification and characterization of viral DNA. J. Virol. 1973, 12, 1473–1481. [Google Scholar]
- Drouet, E.; Michelson, S.; Denoyel, G.; Colimon, R. Polymerase chain reaction detection of human cytomegalovirus in over 2000 blood specimens correlated with virus isolation and related to urinary virus excretion. J. Virol. Methods 1993, 45, 259–276. [Google Scholar] [CrossRef]
- Jacob, R.J.; Morse, L.S.; Roizman, B. Anatomy of herpes simplex virus DNA. XII. Accumulation of head-to-tail concatemers in nuclei of infected cells and their role in the generation of the four isomeric arrangements of viral DNA. J. Virol. 1979, 29, 448–457. [Google Scholar]
- Hughes, T.S.; Langer, S.J.; Johnson, K.W.; Chavez, R.A.; Watkins, L.R.; Milligan, E.D.; Leinwand, L.A. Intrathecal injection of naked plasmid DNA provides long-term expression of secreted proteins. Mol. Ther. 2009, 17, 88–94. [Google Scholar] [CrossRef]
- Kawada, J.I.; Okuno, Y.; Torii, Y.; Okada, R.; Hayano, S.; Ando, S.; Kamiya, Y.; Kojima, S.; Ito, Y. Identification of Viruses in Cases of Pediatric Acute Encephalitis and Encephalopathy Using Next-Generation Sequencing. Sci. Rep. 2016, 6, 33452. [Google Scholar] [CrossRef]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Fragments (n) |
|---|---|
| HSV-1 | 40 1 |
| HSV-2 | 16 |
| VZV | 352 |
| EBV | 129 |
| CMV | 137 |
| HHV-7 | 473 |
| Enterovirus | 22 |
| HIV-1 | 19 |
| VIDISCA-NGS | DNase-free VIDISCA-NGS | |
|---|---|---|
| RNA virus | ||
| Enterovirus | 6/8 1 | 2/8 |
| HIV-1 | 8/10 | 6/10 |
| Total | 14/18 | 8/18 |
| Herpesvirus | ||
| HSV-1/2 | 0/14 | 4/14 |
| VZV | 1/8 | 5/8 |
| EBV | 0/12 | 0/12 |
| CMV | 0/2 | 2/2 |
| HHV-7 | 0/2 | 0/2 |
| Total | 1/38 | 11/38 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edridge, A.W.D.; Deijs, M.; van Zeggeren, I.E.; Kinsella, C.M.; Jebbink, M.F.; Bakker, M.; van de Beek, D.; Brouwer, M.C.; van der Hoek, L. Viral Metagenomics on Cerebrospinal Fluid. Genes 2019, 10, 332. https://doi.org/10.3390/genes10050332
Edridge AWD, Deijs M, van Zeggeren IE, Kinsella CM, Jebbink MF, Bakker M, van de Beek D, Brouwer MC, van der Hoek L. Viral Metagenomics on Cerebrospinal Fluid. Genes. 2019; 10(5):332. https://doi.org/10.3390/genes10050332
Chicago/Turabian StyleEdridge, Arthur W. D., Martin Deijs, Ingeborg E. van Zeggeren, Cormac M. Kinsella, Maarten F. Jebbink, Margreet Bakker, Diederik van de Beek, Matthijs C. Brouwer, and Lia van der Hoek. 2019. "Viral Metagenomics on Cerebrospinal Fluid" Genes 10, no. 5: 332. https://doi.org/10.3390/genes10050332
APA StyleEdridge, A. W. D., Deijs, M., van Zeggeren, I. E., Kinsella, C. M., Jebbink, M. F., Bakker, M., van de Beek, D., Brouwer, M. C., & van der Hoek, L. (2019). Viral Metagenomics on Cerebrospinal Fluid. Genes, 10(5), 332. https://doi.org/10.3390/genes10050332