Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa

 ,
, 
Abstract
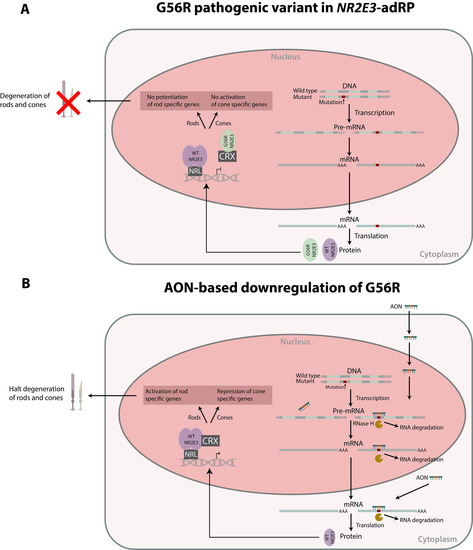
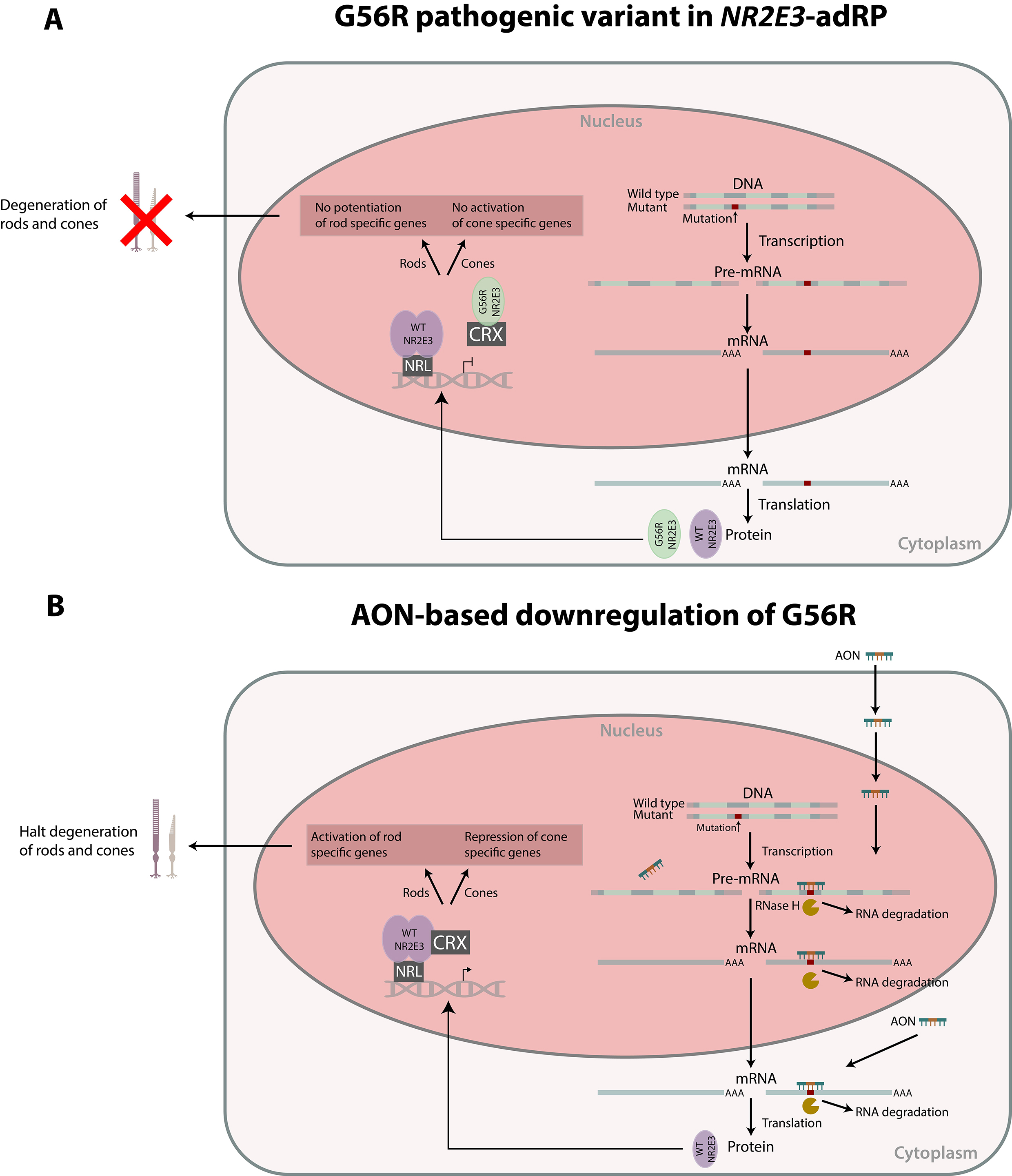
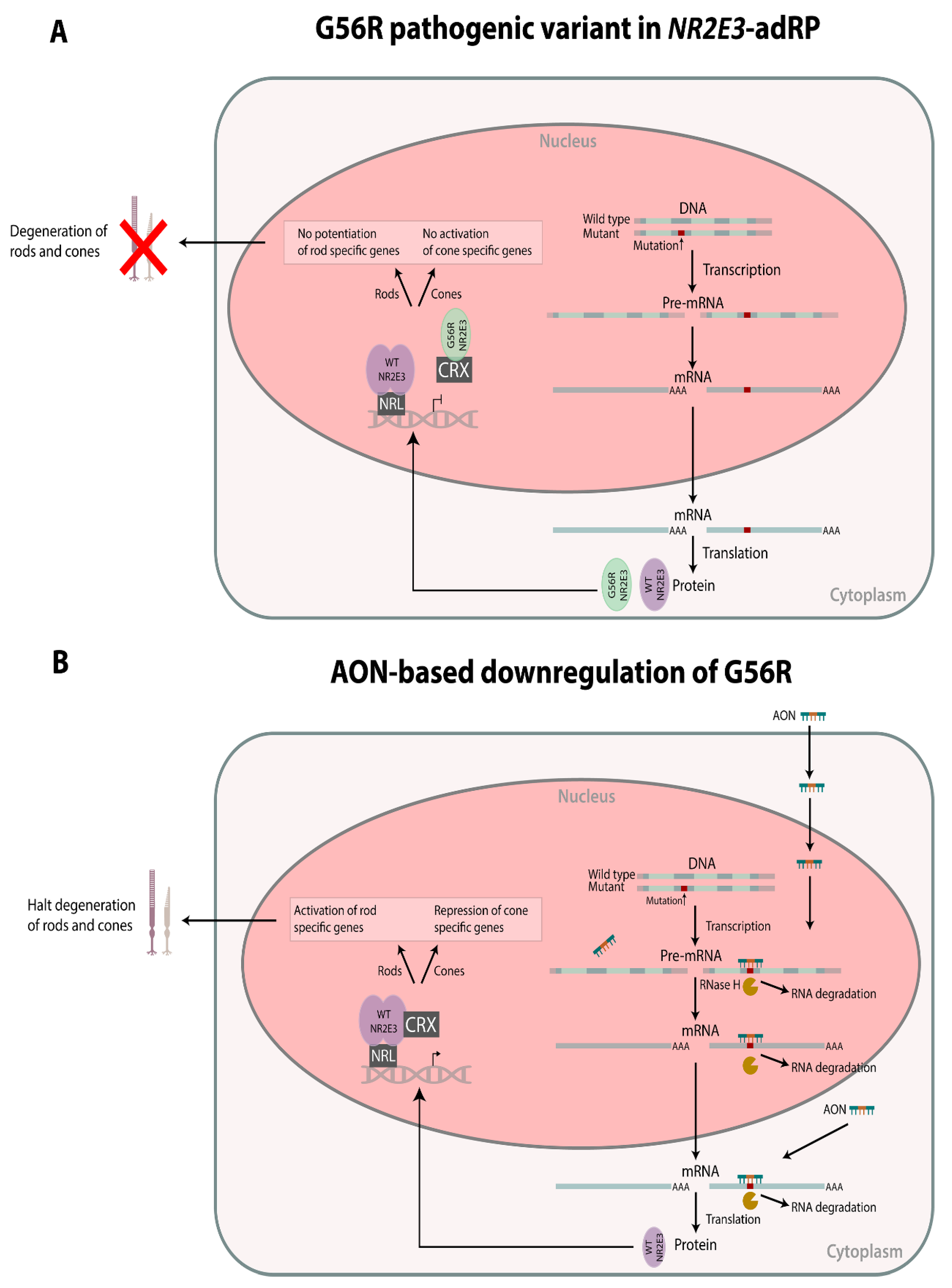
:
1. Introduction
2. Materials and Methods
2.1. AON Design
2.2. Generation of Mutant c.166G>A Expression Construct
2.3. Cell Culture and AON Transfection Experiments
2.4. RNA Isolation and Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.5. Western Blot Analysis
3. Results
3.1. AON Design
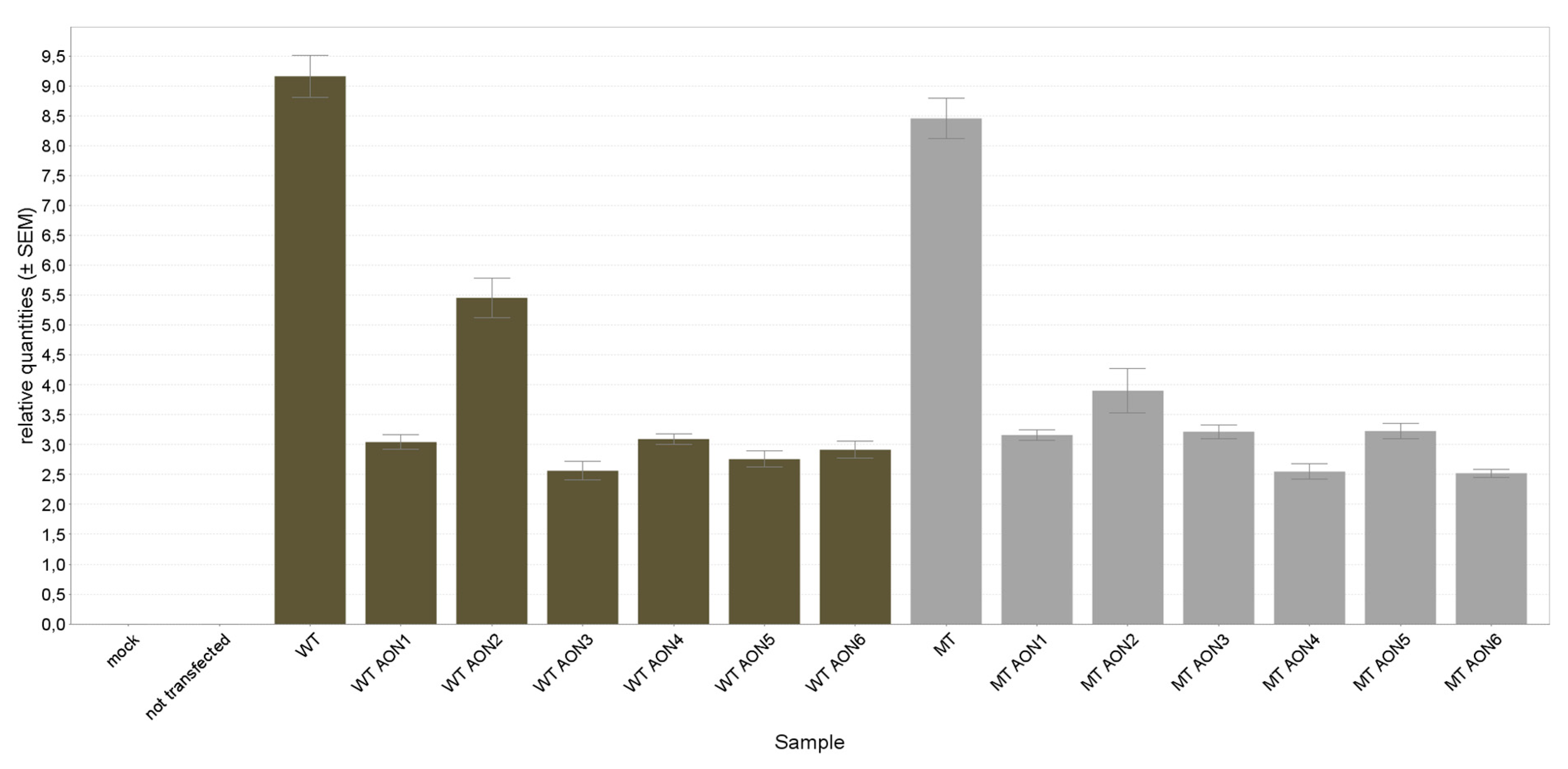
3.2. NR2E3 mRNA Expression Analysis
3.3. NR2E3 Protein Expression Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Den Hollander, A.I.; Black, A.; Bennett, J.; Cremers, F.P. Lighting a candle in the dark: Advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Inverstigation 2010, 120, 3042–3053. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Takezawa, S.; Hara, K.; Yu, R.T.; Umesono, Y.; Agata, K.; Taniwaki, M.; Yasuda, K.; Umesono, K. Identification of a photoreceptor cell-specific nuclear receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 4814–4819. [Google Scholar] [CrossRef]
- Peng, G.H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef]
- Chen, J.; Rattner, A.; Nathans, J. The Rod Photoreceptor-Specific Nuclear Receptor Nr2e3 Represses Transcription of Multiple Cone-Specific Genes. J. Neurosci. 2005, 25, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Aleman, T.S.; Cideciyan, A.V.; Khanna, R.; Samuel, G.; Swaroop, A. In vivo function of the orphan nuclear receptor NR2E3 in establishing photoreceptor identity during mammalian retinal development. Hum. Mol. Genet. 2006, 15, 2588–2602. [Google Scholar] [CrossRef]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; Baere, E. De Recurrent Mutation in the First Zinc Finger of the Orphan Nuclear Receptor NR2E3 Causes Autosomal Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157. [Google Scholar] [CrossRef]
- Van Cauwenbergh, C.; Coppieters, F.; Roels, D.; De Jaegere, S.; Flipts, H.; De Zaeytijd, J.; Walraedt, S.; Claes, C.; Fransen, E.; Van Camp, G.; et al. Mutations in splicing factor genes are a major cause of autosomal dominant retinitis pigmentosa in belgian families. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef]
- Gire, A.I.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-wheaton, D.; John, R.; Daiger, S.P. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2007, 13, 1970–1975. [Google Scholar]
- Blanco-Kelly, F.; García Hoyos, M.; Lopez Martinez, M.A.; Lopez-Molina, M.I.; Riveiro-Alvarez, R.; Fernandez-San Jose, P.; Avila-Fernandez, A.; Corton, M.; Millan, J.M.; García Sandoval, B.; et al. Dominant retinitis pigmentosa, p.Gly56Arg mutation in NR2E3: Phenotype in a large cohort of 24 cases. PLoS ONE 2016, 11, 1–13. [Google Scholar] [CrossRef]
- Escher, P.; Gouras, P.; Roduit, R.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.C.; Hayashi, M.; Zernant, J.; et al. Mutations in NR2E3 Can Cause Dominant or Recessive Retinal Degenerations in the Same Family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef]
- Roduit, R.; Escher, P.; Schorderet, D.F. Mutations in the DNA-Binding Domain of NR2E3 Affect In Vivo Dimerization and Interaction with CRX. PLoS ONE 2009, 4, 1–12. [Google Scholar] [CrossRef]
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef]
- Shibata, S.B.; Ranum, P.T.; Moteki, H.; Pan, B.; Goodwin, A.T.; Goodman, S.S.; Abbas, P.J.; Holt, J.R.; Smith, R.J.H. RNA Interference Prevents Autosomal-Dominant Hearing Loss. Am. J. Hum. Genet. 2016, 98, 1101–1113. [Google Scholar] [CrossRef]
- Gualandi, F.; Manzati, E.; Sabatelli, P.; Passarelli, C.; Bovolenta, M.; Pellegrini, C.; Perrone, D.; Squarzoni, S.; Pegoraro, E.; Bonaldo, P.; et al. Antisense-Induced Messenger Depletion Corrects a COL6A2 Dominant Mutation in Ullrich Myopathy. Hum. Gene Ther. 2012, 23, 1313–1318. [Google Scholar] [CrossRef]
- Bolduc, V.; Zou, Y.; Ko, D.; Bönnemann, C.G. siRNA-mediated Allele-specific Silencing of a COL6A3 Mutation in a Cellular Model of Dominant Ullrich Muscular Dystrophy. Mol. Ther. Nucleic Acids 2014, 3, e147. [Google Scholar] [CrossRef]
- Marrosu, E.; Ala, P.; Muntoni, F.; Zhou, H. Gapmer Antisense Oligonucleotides Suppress the Mutant Allele of COL6A3 and Restore Functional Protein in Ullrich Muscular Dystrophy. Mol. Ther. Nucleic Acids 2017, 8, 416–427. [Google Scholar] [CrossRef]
- Trochet, D.; Prudhon, B.; Beuvin, M.; Peccate, C.; Lorain, S.; Julien, L.; Benkhelifa-Ziyyat, S.; Rabai, A.; Mamchaoui, K.; Ferry, A.; et al. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. EMBO Mol. Med. 2017, 10, 239–253. [Google Scholar] [CrossRef]
- Murray, S.F.; Jazayeri, A.; Matthes, M.T.; Yasumura, D.; Yang, H.; Peralta, R.; Watt, A.; Freier, S.; Hung, G.; Adamson, P.S.; et al. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6362–6375. [Google Scholar] [CrossRef]
- Jiang, L.; Frederick, J.M.; Baehr, W. RNA interference gene therapy in dominant retinitis pigmentosa and cone-rod dystrophy mouse models caused by GCAP1 mutations. Front. Mol. Neurosci. 2014, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, M.E.; Southwell, A.L.; Kordasiewicz, H.; Watt, A.T.; Skotte, N.H.; Doty, C.N.; Vaid, K.; Villanueva, E.B.; Swayze, E.E.; Bennett, C.F.; et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013, 41, 9634–9650. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Skotte, N.H.; Kordasiewicz, H.B.; Østergaard, M.E.; Watt, A.T.; Carroll, J.B.; Doty, C.N.; Villanueva, E.B.; Petoukhov, E.; Vaid, K.; et al. In Vivo Evaluation of Candidate Allele-specific Mutant Huntingtin Gene Silencing Antisense Oligonucleotides. Mol. Ther. Am. Soc. Gene Cell Ther. 2014, 22, 2093–2106. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2018, 25, 225–228. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; Peters, T.A.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Acids 2016, 5, e381. [Google Scholar] [CrossRef] [PubMed]
- Slijkerman, R.W.; Goloborodko, A.; Broekman, S.; de Vrieze, E.; Hetterschijt, L.; Peters, T.A.; Gerits, M.; Kremer, H.; Van Wijk, E. Poor Splice-Site Recognition in a Humanized Zebrafish Knockin Model for the Recurrent Deep-Intronic c.7595-2144A>G Mutation in USH2A. Zebrafish 2018, 15, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef]
- Garanto, A.; van der Velde-Visser, S.D.; Cremers, F.P.M.; Collin, R.W.J. Antisense Oligonucleotide-Based Splice Correction of a Deep-Intronic Mutation in CHM Underlying Choroideremia. In Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; pp. 83–89. [Google Scholar]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.; Bauwens, M.; Bax, N.; Van den Born, I.; Verheij, J.; Pott, J.-W.; Thiadens, A.; et al. Deep-intronic ABCA4 mutations explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel non-coding splice, cis- regulatory, structural and recurrent hypomorphic variants. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT -PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef]
- Schneier, A.J.; Fulton, A.B. The hermansky-pudlak syndrome: Clinical features and imperatives from an ophthalmic perspective. Semin. Ophthalmol. 2013, 28, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Grünweller, A.; Hartmann, R.K. Locked Nucleic Acid Oligonucleotides: The next generation of antisense agents? BioDrugs 2007, 21, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.; Bentzen, J.; Meldgaard, M.; Jakobsen, M.H.; Fenger, M.; Kauppinen, S.; Skouv, J. LNA-enhanced detection of single nucleotide polymorphisms in the apolipoprotein E. Nucleic Acids Res. 2002, 30, e100. [Google Scholar] [CrossRef] [PubMed]
- Schorderet, D.F.; Escher, P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum. Mutat. 2009, 30, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Terray, A.; Slembrouck, A.; Nanteau, C.; Chondroyer, C.; Zeitz, C.; Sahel, J.-A.; Audo, I.; Reichman, S.; Goureau, O. Generation of an induced pluripotent stem cell (iPSC) line from a patient with autosomal dominant retinitis pigmentosa due to a mutation in the NR2E3 gene. Stem Cell Res. 2017, 24, 1–4. [Google Scholar] [CrossRef]
- Tucker, B.; Mullins, R.F.; Streb, L.M.; Anfinson, K.; Eyestone, M.E.; Kaalberg, E.; Riker, M.J.; Drack, A.V.; Braun, T.; Stone, E.M. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife 2013, 2, e00824. [Google Scholar] [CrossRef]
- Jepsen, J.S.; Pfundheller, H.M.; Lykkesfeldt, A.E. Downregulation of p21(WAF1/CIP1) and Estrogen Receptor? in MCF-7 Cells by Antisense Oligonucleotides Containing Locked Nucleic Acid (LNA). Oligonucleotides 2004, 14, 147–156. [Google Scholar] [CrossRef]
- Ten Asbroek, A.L.; Fluiter, K.; van Groenigen, M.; Nooij, M.; Baas, F. Polymorphisms in the large subunit of human RNA polymerase II as target for allele-specific inhibition. Nucleic Acids Res. 2000, 28, 1133–1138. [Google Scholar] [CrossRef]
- Monia, B.P.; Johnston, J.F.; Ecker, D.J.; Zounes, M.A.; Lima, W.F.; Freier, S.M. Selective inhibition of mutant Ha-ras mRNA expression by antisense oligonucleotides. J. Biol. Chem. 1992, 267, 19954–19962. [Google Scholar]
- Magner, D.; Biala, E.; Lisowiec-Wachnicka, J.; Kierzek, R. Influence of mismatched and bulged nucleotides on SNP-preferential RNase H cleavage of RNA-antisense gapmer heteroduplexes. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gapmers | Sequence (5′→3′) | Complete Match Bowtie | Complete Match Bowtie2 |
|---|---|---|---|
| AON1 AON2 | Mg*Mu*Mg*Mc*Mu*t*c*c*T*g*c*t*g*c*t*Mg*Mc*Mu*Mg*Mu Mg*Mu*Mg*Mc*Mu*t*c*c*LT*g*c*t*g*c*t*Mg*Mc*Mu*Mg*Mu | gDNA: no cDNA: SLC29A4P1 | gDNA: no cDNA: no |
| Mg*Mc*Mu*t*c*c*T*g*c*t*g*c*t*Mg*Mc*Mu Mg*Mc*Mu*t*c*c*LT*g*c*t*g*c*t*Mg*Mc*Mu | gDNA: ICOS cDNA: GPR27 | gDNA: CACNA2D3 cDNA: PCDHAC2 | |
| Mg*Mu*Mg*Mc*Mu*t*c*c*T*g*c*t*g*Mc*Mu*Mg*Mc*Mu Mg*Mu*Mg*Mc*Mu*t*c*c*LT*g*c*t*g*Mc*Mu*Mg*Mc*Mu | gDNA: intergenicc DNA: HCG9P5 | gDNA: intergenicc DNA: PRDM8 | |
| AON3 AON4 | Mc*Ma*Mu*Ma*Mg*t*g*c*t*t*c*c*T*g*c*Mu*Mg*Mc*Mu*Mg Mc*Ma*Mu*Ma*Mg*t*g*c*t*t*c*c*LT*g*c*Mu*Mg*Mc*Mu*Mg | gDNA: no cDNA: no | gDNA: no cDNA: no |
| Mu*Ma*Mg*t*g*c*t*t*c*c*T*g*c*Mu*Mg*Mc Mu*Ma*Mg*t*g*c*t*t*c*c*LT*g*c*Mu*Mg*Mc | gDNA: intergenicc DNA: HCG9P5 | gDNA: TANGO6 cDNA: HCG9P5 | |
| Ma*Mu*Ma*Mg*Mu*g*c*t*t*c*c*T*g*Mc*Mu*Mg*Mc*Mu Ma*Mu*Ma*Mg*Mu*g*c*t*t*c*c*LT*g*Mc*Mu*Mg*Mc*Mu | gDNA: AC007908.1 cDNA: no | gDNA: no cDNA: HCG9P5 | |
| AON5 AON6 | Mc*Mu*Mu*Mc*Mc*T*g*c*t*g*c*t*g*c*t*Mg*Mu*Mc*Mu*Mc Mc*Mu*Mu*Mc*Mc*LT*g*c*t*g*c*t*g*c*t*Mg*Mu*Mc*Mu*Mc | gDNA: no cDNA: PCDHAC2 | gDNA: no cDNA: ACVR2B |
| Mu*Mu*Mc*c*T*g*c*t*g*c*t*g*c*Mu*Mg*Mu Mu*Mu*Mc*c*LT*g*c*t*g*c*t*g*c*Mu*Mg*Mu | gDNA: EIF3H cDNA: LMAN2 | gDNA: PDCD1LG2 cDNA: DYRK1A | |
| Mc*Mu*Mu*Mc*Mc*T*g*c*t*g*c*t*g*Mc*Mu*Mg*Mu*Mc Mc*Mu*Mu*Mc*Mc*LT*g*c*t*g*c*t*g*Mc*Mu*Mg*Mu*Mc | gDNA: SEMA5A cDNA: LY6H | gDNA: SEMA5A cDNA: PLXNB3 |
| AON | Expression WT NR2E3 (%) | SEM (%) | Expression MT NR2E3 (%) | SEM (%) |
|---|---|---|---|---|
| NT | 100.00 | 3.84 | 100.00 | 4.01 |
| AON1 | 33.22 | 4.06 | 37.37 | 2.86 |
| AON2 | 59.55 | 6.12 | 46.15 | 9.65 |
| AON3 | 27.98 | 5.98 | 38.04 | 3.51 |
| AON4 | 33.76 | 2.94 | 30.15 | 4.89 |
| AON5 | 30.10 | 4.95 | 38.16 | 3.87 |
| AON6 | 31.81 | 4.94 | 29.89 | 4.26 |
| AON | Expression WT NR2E3 (%) | Expression MT NR2E3 (%) |
|---|---|---|
| NT | 100.00 | 100.00 |
| AON1 | 22.87 | 19.33 |
| AON2 | 55.55 | 58.85 |
| AON3 | 27.22 | 25.45 |
| AON4 | 70.24 | 52.47 |
| AON5 | 45.08 | 8.88 |
| AON6 | 27.52 | 21.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naessens, S.; Ruysschaert, L.; Lefever, S.; Coppieters, F.; De Baere, E. Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes 2019, 10, 363. https://doi.org/10.3390/genes10050363
Naessens S, Ruysschaert L, Lefever S, Coppieters F, De Baere E. Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes. 2019; 10(5):363. https://doi.org/10.3390/genes10050363
Chicago/Turabian StyleNaessens, Sarah, Laurien Ruysschaert, Steve Lefever, Frauke Coppieters, and Elfride De Baere. 2019. "Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa" Genes 10, no. 5: 363. https://doi.org/10.3390/genes10050363
APA StyleNaessens, S., Ruysschaert, L., Lefever, S., Coppieters, F., & De Baere, E. (2019). Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes, 10(5), 363. https://doi.org/10.3390/genes10050363