RNA Sequencing Analysis of Chicken Cecum Tissues Following Eimeria tenella Infection in Vivo

Abstract
:1. Introduction
2. Materials and Methods
2.1. E. tenella Infection in Chickens and Tissue Collection
2.2. RNA Isolation and Quality Assessment
2.3. Library Preparation and Transcriptome Sequencing
2.4. Sequence Read Mapping to the Gallus Gallus Reference Genome
2.5. Quantification and Differential Expression Analysis of Transcripts
2.6. GO and KEGG Enrichment Analysis of Differentially Expressed Genes
2.7. Validation of Differentially Expressed Genes by qRT-PCR
3. Results
3.1. Clinical Observation
3.2. RNA-Seq Data Analysis
3.3. Identification of Differentially Expressed Genes in Chicken Ceca upon E. tenella Infection
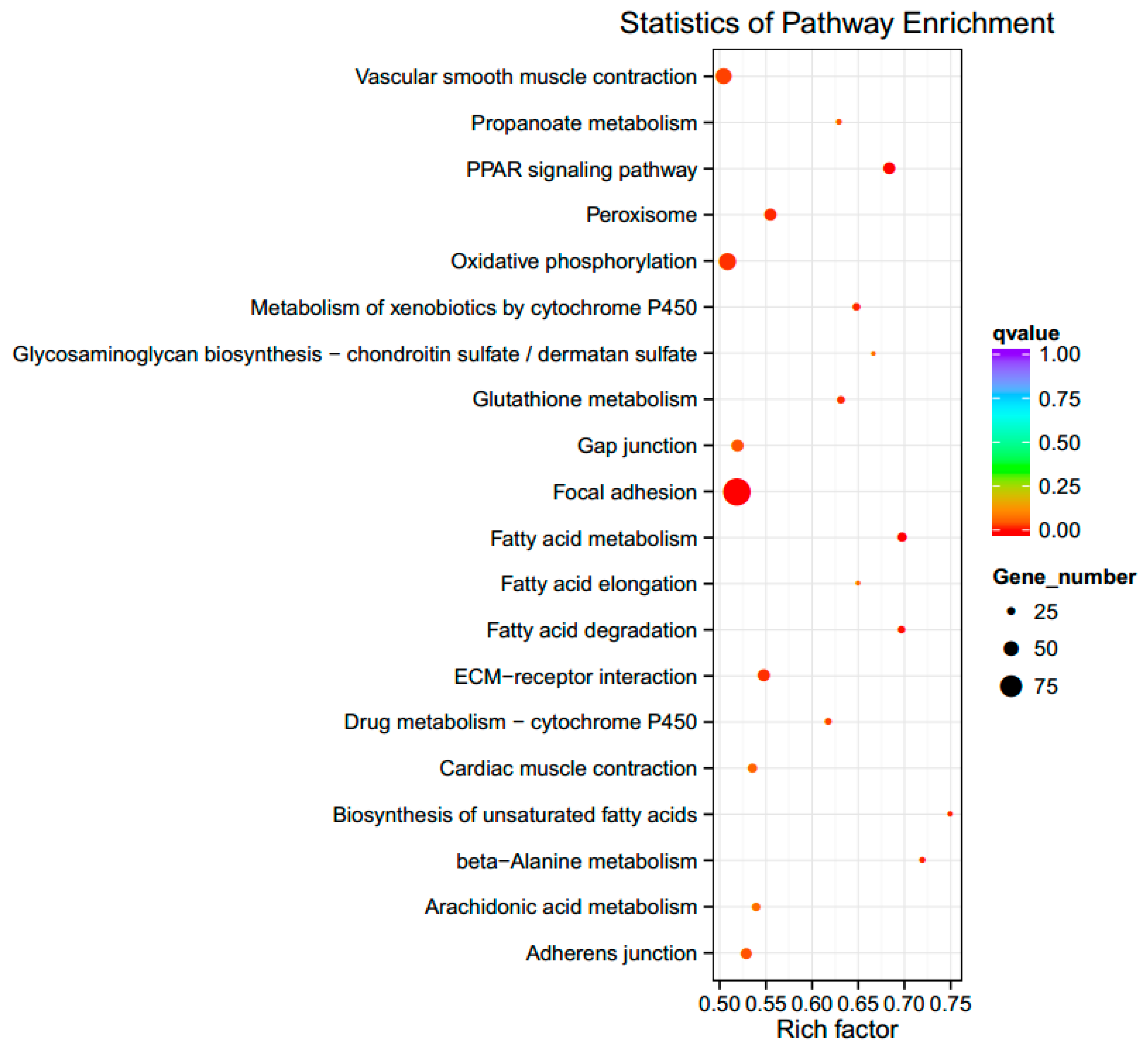
3.4. GO Enrichment and KEGG Pathway Analysis for DEGs
3.5. Real-Time PCR Validation of Differential Gene Expression in Chicken Ceca
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blake, D.P.; Tomley, F.M. Securing poultry production from the ever-present Eimeria challenge. Trends Parasitol. 2014, 30, 12–19. [Google Scholar] [CrossRef]
- Chapman, H.D.; Jeffers, T.K.; Williams, R.B. Forty years of monensin for the control of coccidiosis in poultry. Poult. Sci. 2010, 89, 1788–1801. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.I.; Lillehoj, H.S.; Lee, S.H.; Lee, K.W.; Park, M.S.; Bauchan, G.R.; Lillehoj, E.P.; Bertrand, F.; Dupuis, L.; Deville, S. Immunoenhancing effects of montanide ISA oil-based adjuvants on recombinant Coccidia antigen vaccination against Eimeria acervulina infection. Vet. Parasitol. 2010, 172, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.S.; Wang, Y.Y.; Liao, S.Q.; Wu, C.Y.; Lv, M.N.; Li, J.; Tong, Z.X.; Sun, M.F. Partial protective of chickens against Eimeria tenella challenge with recombinant EtMIC-1 antigen. Parasitol. Res. 2013, 112, 2281–2287. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, J.; Li, J.; Gong, P.; Zhang, X. Protective immunity induced by a DNA vaccine encoding Eimeria tenellar homboid against homologous challenge. Parasitol. Res. 2013, 112, 251–257. [Google Scholar] [CrossRef]
- Mathis, G.; Waldrip, D.; Dickson, J.; Cookson, K.; LaVorgna, M.; Schaeffer, J. Effect of lasalocid or salinomycin administration on performance and immunity following Coccidia vaccination of commercial broilers. J. Appl. Poult. Res. 2014, 23, 577–585. [Google Scholar] [CrossRef]
- Lillehoj, H.S.; Hong, Y.; Kim, C. Quantitative genetic and functional genomics approaches to investigating parasite disease resistance and protective immune mechanisms in avian coccidiosis. Dev. Biol. (Basel) 2008, 132, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Cai, J.; Gong, W.; Yan, H.; Luo, X.; Tian, G.; Zhang, S.; Zhang, H.; Zhu, G.; Cai, X. Transcriptome analysis in chicken cecal epithelia upon infection by Eimeria tenella in vivo. PLoS ONE 2013, 8, e64236. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Lillehoj, H.; Min, W.; Kim, C.H.; Park, M.S.; Hong, Y.H.; Lillehoj, E.P. Comparative microarray analysis of intestinal lymphocytes following Eimeria acervulina, E. maxima, or E. tenella infection in the chicken. PLoS ONE 2011, 6, e27712. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Woods, W.G.; Richards, D.G.; Gasser, R.B. Investigating a persistent coccidiosis problem on a commercial broiler-breeder farm utilising PCR-coupled capillary electrophoresis. Parasitol. Res. 2007, 101, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Long, P.L.; Johnson, J.; Wyatt, R.D. Pathological and clinical effects of Eimeria tenella in partially immune chickens. J. Comp. Pathol. 1981, 91, 581–587. [Google Scholar] [CrossRef]
- Jiao, J.; Yang, Y.; Liu, M.; Li, J.; Cui, Y.; Yin, S.; Tao, J. Artemisinin and Artemisia annua leaves alleviate Eimeria tenella infection by facilitating apoptosis of host cells and suppressing inflammatory response. Vet. Parasitol. 2018, 254, 172–177. [Google Scholar] [CrossRef] [PubMed]
- She, R.; Fei, C.; Chen, H.; Wang, X.; Wang, M.; Zhang, K.; Zhang, L.; Wang, C.; Liu, Y.; Zheng, W.; et al. Action of nitromezuril against Eimeria tenella with clinically anticoccidial indices and histopathology. Parasitol. Res. 2017, 116, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq: A python framework to work with high-throughput sequencing data. Bioinformatics 2010, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.J.; Lillehoj, H.S.; Allen, P.C.; Van Tassell, C.P.; Sonstegard, T.S.; Cheng, H.H.; Pollock, D.; Sadjadi, M.; Min, W.; Emara, M.G. Mapping quantitative trait loci associated with resistance to coccidiosis and growth. Poult. Sci. 2003, 82, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Lei, X.W.; Yang, F.L.; Li, M.Y.; Tang, C. Reference gene selection for normalization of PCR analysis in chicken embryo fibroblast infected with H5N1 AIV. Virol. Sin. 2010, 25, 425–431. [Google Scholar] [CrossRef]
- Kim, C.H.; Lillehoj, H.S.; Hong, Y.H.; Keeler, C.L., Jr.; Lillehoj, E.P. Comparison of global transcriptional responses to primary and secondary Eimeria acervulina infections in chickens. Dev. Comp. Immunol. 2010, 34, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Fiorucci, S. PPARs and other nuclear receptors in inflammation. Curr. Opin. Pharmacol. 2006, 6, 421–427. [Google Scholar] [CrossRef]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: molecular mechanisms and pathophysiological relevance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Pinard, M.H.; Gay, C.; Pastoret, P.P.; Dodet, B. Animal genomics for animal health. Dev. Biol. (Basel) 2008, 132, 73. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, J.; Weng, X.; Liu, F.; He, L.; Zhou, D.; Liu, Y. Comparative transcriptome analysis reveals that the extracellular matrix receptor interaction contributes to the venous metastases of hepatocellular carcinoma. Cancer Genet. 2015, 208, 482–491. [Google Scholar] [CrossRef]
- Lan, G.; Xie, W.; Li, L.; Zhang, M.; Liu, D.; Tan, Y.L.; Cheng, H.P.; Gong, D.; Huang, C.; Zheng, X.L.; et al. MicroRNA-134 actives lipoprotein lipase-mediated lipid accumulation and inflammatory response by targeting angiopoietin-like 4 in THP-1 macrophages. Biochem. Biophys. Res. Commun. 2016, 472, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Gealekman, O.; Burkart, A.; Chouinard, M.; Nicoloro, S.M.; Straubhaar, J.; Corvera, S. Enhanced angiogenesis in obesity and in response to PPARgamma activators through adipocyte VEGF and ANGPTL4 production. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1056–E1064. [Google Scholar] [CrossRef]
- Reinartz, A.; Ehling, J.; Leue, A.; Liedtke, C.; Schneider, U.; Kopitz, J.; Weiss, T.; Hellerbrand, C.; Weiskirchen, R.; Knuchel, R.; et al. Lipid-induced up-regulation of human acyl-CoA synthetase 5 promotes hepatocellular apoptosis. Biochim. Biophys. Acta 2010, 1801, 1025–1035. [Google Scholar] [CrossRef]
- Catala-Rabasa, A.; Ndagire, D.; Sabio, J.M.; Fedetz, M.; Matesanz, F.; Alcina, A. High ACSL5 transcript levels associate with systemic lupus erythematosus and apoptosis in Jurkat T lymphocytes and peripheral blood cells. PLoS ONE 2011, 6, e28591. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- DeGrendele, H.C.; Estess, P.; Siegelman, M.H. Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science 1997, 278, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Sakima, M.; Hayashi, H.; Mamun, A.A.; Sato, M. VEGFR-3 signaling is regulated by a G-protein activator, activator of G-protein signaling 8, in lymphatic endothelial cells. Exp. Cell Res. 2018, 368, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, C.; Detmar, M. Pathways targeting tumor lymphangiogenesis. Clin. Cancer Res. 2006, 12, 6865–6868. [Google Scholar] [CrossRef]
- Ying, J.; Li, H.; Cui, Y.; Wong, A.H.; Langford, C.; Tao, Q. Epigenetic disruption of two proapoptotic genes MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas through hypermethylation of a common bidirectional promoter. Leukemia 2006, 20, 1173–1175. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene | Forward primers (5′-3′) | Reverse primers (5′-3′) | Length (bp) | GenBank ID |
|---|---|---|---|---|
| ANPEP | GCCCACCTGGGACATTAAAGA | GATCTGTGCTGGGGTGTTGA | 127 | NM_204861.1 |
| HKDC1 | AAAACCCACCCCCAATACCC | CCAGGGTTAGCACGAAAGGT | 193 | XM_421579.4 |
| CA7 | GAGGCCGCCAATGGAAGAG | CACTCCGAAGGTCCCGC | 102 | XM_004944247.1 |
| BEST4 | GATTCCCTGCGTCTGGTTCA | AATGGTAACCACCTGCGTGT | 181 | NM_001252125.1 |
| OTOP2 | GTGGCTCCTGAAAGTGGGAA | CCCCACGTTCTTCCACATGA | 166 | XM_003642368.2 |
| COL8A1 | CAGTTGTTTCGCACCCAAGG | ACTCTGTTCTCAGTCGCTGT | 187 | NM_001293134.1 |
| TAC1 | CGTCCGTCCATCAGTGTGTT | CAGCTCCTCCTTCTGCTCG | 180 | XM_004939318.1 |
| CHRDL2 | TGAACCCAAAACGGCCAGAT | TAGCACCTCGTGTTGCCATT | 145 | XM_004939009.1 |
| CEBPB | CTCCTACCTGGGCTACCAGT | TTGTACTCGTCGCTGTGCTT | 195 | NM_205253.2 |
| CTSL2 | AAAGACCAGGGTCAGTGTGG | TTGATTTCCTTCTGGGCGGG | 141 | NM_001168009.1 |
| YWHAZ | GTTCCCTTGCAAAAACGGCTT | AGACGGAAGTTGGAAGGCTG | 194 | NM_001031343.1 |
| TBP | GAACCACACCTCTGTACCCG | GCAGCAAAACGCTTGGGATT | 196 | NM_205103.1 |
| Sample | Raw Reads | Clean Reads | Total Mapped | Uniquely Mapped | Q20 (%) | GC (%) |
|---|---|---|---|---|---|---|
| JS1 | 64,910,186 | 60,434,754 | 48,799,256 | 48,250,954 | 96.01 | 49.18 |
| JS2 | 52,432,520 | 49,658,574 | 39,706,701 | 39,238,872 | 96.49 | 50.16 |
| JS3 | 61,872,638 | 58,613,210 | 46,465,172 | 45,966,122 | 96.52 | 50.60 |
| JC1 | 63,030,840 | 60,429,518 | 49,847,352 | 48,938,857 | 97.18 | 50.62 |
| JC2 | 60,667,018 | 57,459,296 | 47,046,823 | 46,281,102 | 96.67 | 50.25 |
| JC3 | 64,190,770 | 61,549,834 | 51,224,793 | 50,430,317 | 97.26 | 49.75 |
| Pathway Name | Pathway ID | DEG Number | q-value |
|---|---|---|---|
| PPAR signaling | gga03320 | 24 | 0.003765846 |
| Focal adhesion | gga04510 | 55 | 0.009483415 |
| ECM–receptor interaction | gga04512 | 24 | 0.079280157 |
| Gene | Gene ID | RNA-seq log2(Fold Change) | qRT-PCR log2(Fold Change) |
|---|---|---|---|
| OTOP2 | ENSGALG00000007791 | −6.3318 | −6.1629 |
| CA7 | ENSGALG00000005178 | −5.6135 | −5.4033 |
| HKDC1 | ENSGALG00000021039 | −4.4784 | −3.8074 |
| BEST4 | ENSGALG00000010126 | −4.3859 | −3.9561 |
| ANPEP | ENSGALG00000027501 | −3.9646 | −3.7506 |
| CTSL2 | ENSGALG00000012610 | +2.0931 | +2.2265 |
| CEBPB | ENSGALG00000008014 | +2.3416 | +3.2373 |
| TAC1 | ENSGALG00000009737 | +3.0046 | +4.0644 |
| COL8A1 | ENSGALG00000015253 | +3.7752 | +4.4074 |
| CHRDL2 | ENSGALG00000017308 | +5.2340 | +3.7442 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zou, W.; Yu, H.; Lin, Y.; Dai, G.; Zhang, T.; Zhang, G.; Xie, K.; Wang, J.; Shi, H. RNA Sequencing Analysis of Chicken Cecum Tissues Following Eimeria tenella Infection in Vivo. Genes 2019, 10, 420. https://doi.org/10.3390/genes10060420
Wang X, Zou W, Yu H, Lin Y, Dai G, Zhang T, Zhang G, Xie K, Wang J, Shi H. RNA Sequencing Analysis of Chicken Cecum Tissues Following Eimeria tenella Infection in Vivo. Genes. 2019; 10(6):420. https://doi.org/10.3390/genes10060420
Chicago/Turabian StyleWang, Xiaohui, Wenbin Zou, Hailiang Yu, Yuxin Lin, Guojun Dai, Tao Zhang, Genxi Zhang, Kaizhou Xie, Jinyu Wang, and Huiqiang Shi. 2019. "RNA Sequencing Analysis of Chicken Cecum Tissues Following Eimeria tenella Infection in Vivo" Genes 10, no. 6: 420. https://doi.org/10.3390/genes10060420
APA StyleWang, X., Zou, W., Yu, H., Lin, Y., Dai, G., Zhang, T., Zhang, G., Xie, K., Wang, J., & Shi, H. (2019). RNA Sequencing Analysis of Chicken Cecum Tissues Following Eimeria tenella Infection in Vivo. Genes, 10(6), 420. https://doi.org/10.3390/genes10060420