Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Analyses
2.2. Mutant PAH Recombinant Constructs Preparation
2.3. Human PAH Expression in Escherichia coli and Protein Purification
2.4. PAH Functional Assay
2.5. Eukaryotic Expression
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
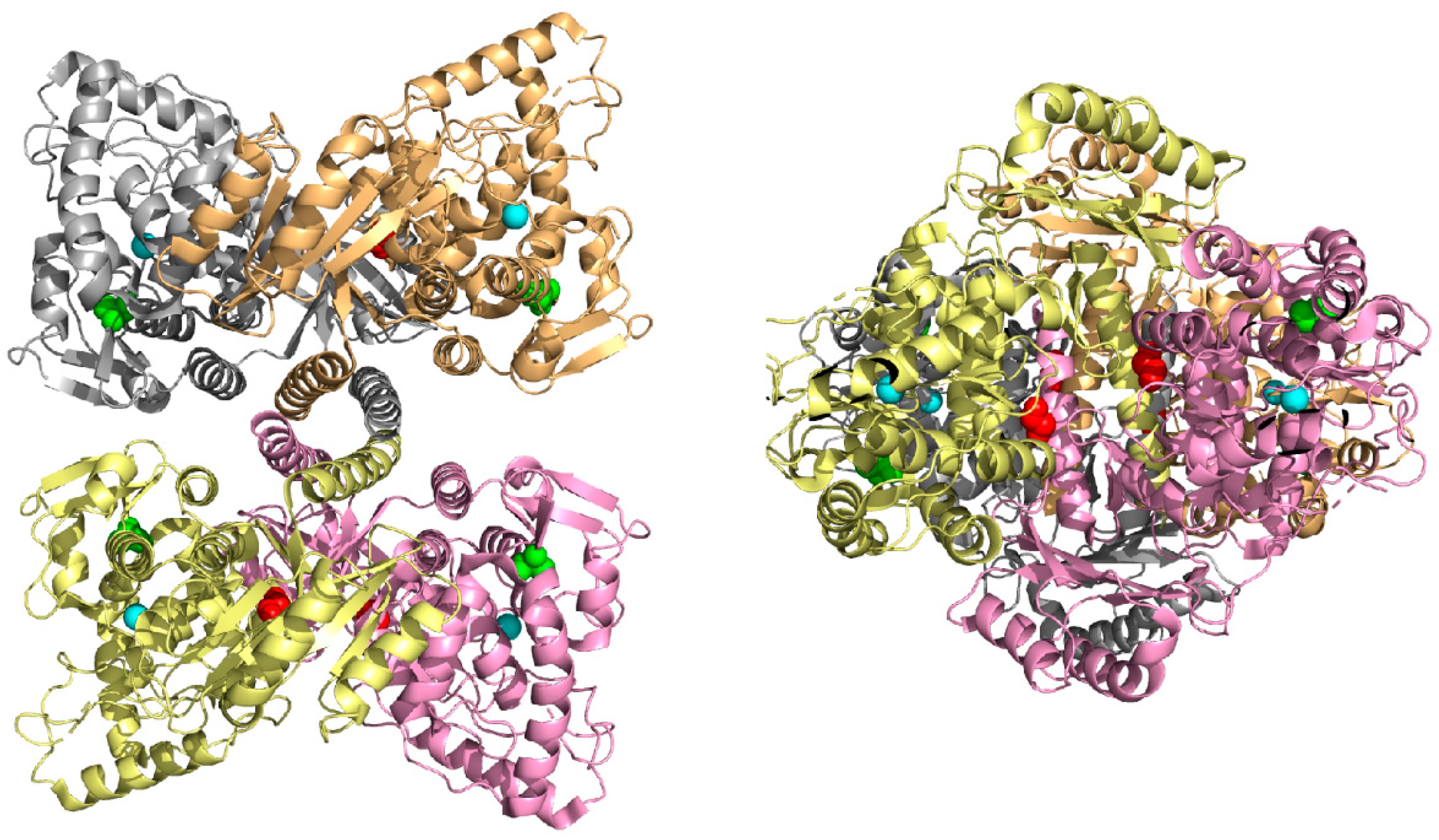
3.1. In Silico Analysis
3.2. Human PAH Expression in Escherichia coli and Oligomeric Pattern
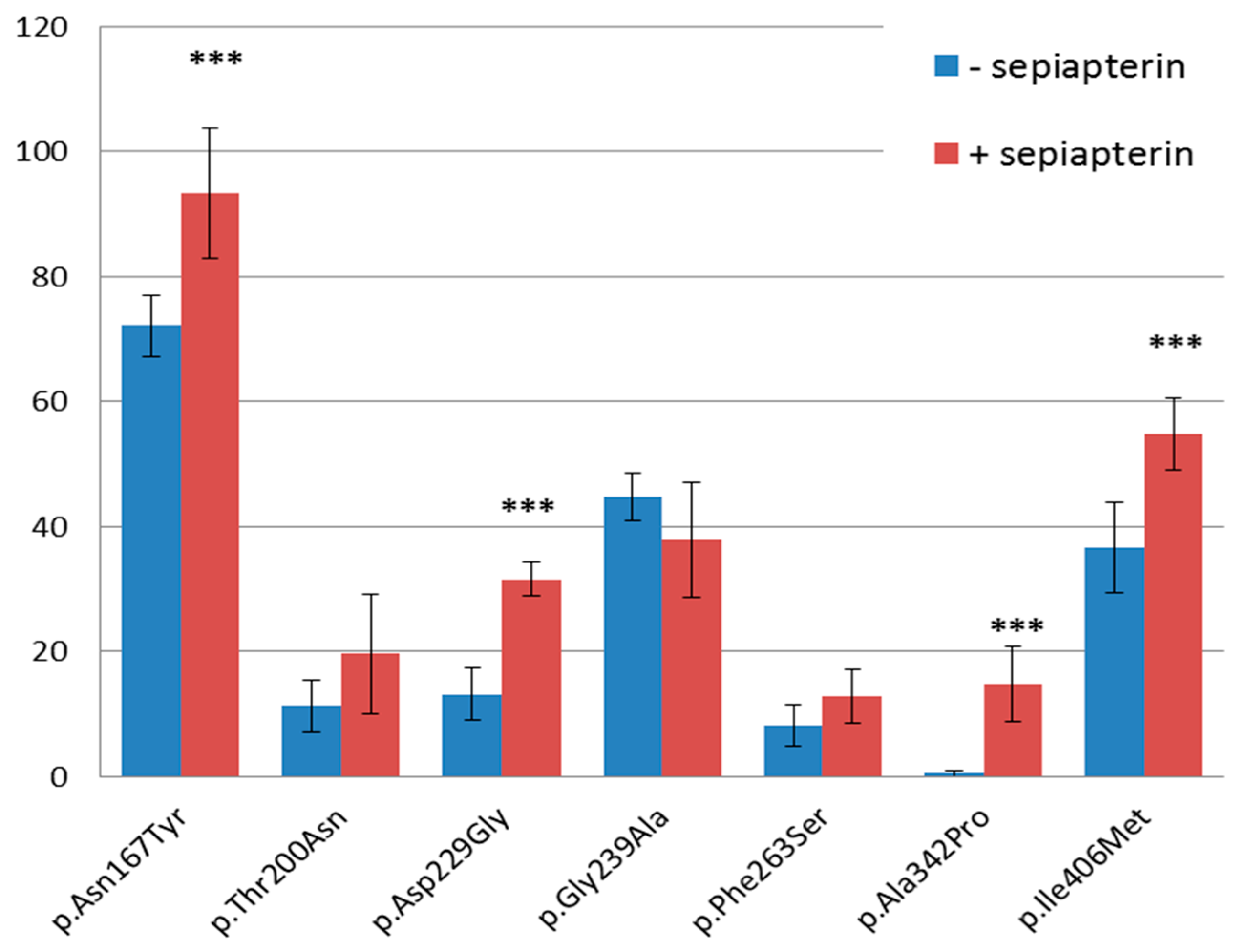
3.3. PAH Functional Assay
3.4. Western Blot Analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loeber, J.G. Neonatal screening in Europe; The situation in 2004. J. Inherit. Metab. Dis. 2007, 30, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Lysinová, M.; Knapková, M.; Dluholucký, S.; Králinský, K. Novorodenecký skríning v súčasnosti. Pediatr. Prax. 2015, 16, 188–191. [Google Scholar]
- Neonatal Screening in the Czech Republic. Available online: https://www.novorozeneckyscreening.cz/en (accessed on 3 April 2019).
- Guldberg, P.; Rey, F.; Zschocke, J.; Romano, V.; Baudouin, F.; Michiels, L.; Ullrich, K.; Hoffmann, G.F.; Burgard, P.; Schmidt, H.; et al. A European multicenter study of phenylalanine hydroxylase deficiency: Classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am. J. Hum. Genet. 1998, 63, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; Hennermann, J.B.; Langenbeck, U.; Lichter-Konecki, U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol. Genet. Metab. 2011, 104, S2–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, B.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.A.; Mamotte, C.D.S.; Burnett, J.R. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin. Biochem. Rev. 2008, 29, 31–41. [Google Scholar] [PubMed]
- Kobe, B.; Jennings, I.G.; House, C.M.; Michell, B.J.; Goodwill, K.E.; Santarsiero, B.D.; Stevens, R.C.; Cotton, R.G.; Kemp, B.E. Structural basis of autoregulation of phenylalanine hydroxylase. Nat. Struct. Biol. 1999, 6, 442–448. [Google Scholar] [CrossRef]
- PAHvdb: Phenylalanine Hydroxylase Gene Locus-Specific Database. Available online: http://www.biopku.org/home/pah.asp (accessed on 03 April 2019).
- Pey, A.L.; Stricher, F.; Serrano, L.; Martinez, A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am. J. Hum. Genet. 2007, 81, 1006–1024. [Google Scholar] [CrossRef]
- Scheller, R.; Stein, A.; Nielsen, S.V.; Marin, F.I.; Gerdes, A.M.; Di Marco, M.; Papaleo, E.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Toward mechanistic models for genotype-phenotype correlations in phenylketonuria using protein stability calculations. Hum. Mutat. 2019, 40, 444–457. [Google Scholar] [CrossRef]
- Bernegger, C.; Blau, N. High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: A Study of 1919 patients observed from 1988 to 2002. Mol. Genet. Metab. 2002, 77, 304–313. [Google Scholar] [CrossRef]
- Burton, B.K.; Bausell, H.; Katz, R.; Laduca, H.; Sullivan, C. Sapropterin therapy increases stability of blood phenylalanine levels in patients with BH4-responsive phenylketonuria (PKU). Mol. Genet. Metab. 2010, 101, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Elsas, L.J.; Greto, J.; Wierenga, A. The effect of blood phenylalanine concentration on KuvanTM response in Phenylketonuria. Mol. Genet. Metab. 2011, 102, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.L.; Milanowski, A.; Chakrapani, A.; Cleary, M.; Lee, P.; Trefz, F.K.; Whitley, C.B.; Feillet, F.; Feigenbaum, A.S.; Bebchuk, J.D.; et al. Sapropterin research group. Efficacy of Sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: A phase III randomised placebo-controlled study. Lancet 2007, 370, 504–510. [Google Scholar] [CrossRef]
- Réblová, K.; Hrubá, Z.; Procházková, Z.; Pazdírková, R.; Pouchlá, S.; Zeman, J.; Fajkusová, L. Hyperphenylalaninemia in the Czech Republic: Genotype-phenotype correlations and in silico analysis of novel missense mutations. Clin. Chim. Acta 2013, 419, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Altman, R.B.; Bromberg, Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. 2013, 14, S2. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Réblová, K.; Kulhánek, P.; Fajkusová, L. Computational study of missense mutations in phenylalanine hydroxylase. J. Mol. Model. 2015, 21, 70. [Google Scholar] [CrossRef]
- Arturo, E.C.; Gupta, K.; Héroux, A.; Stith, L.; Cross, P.J.; Parker, E.J.; Loll, P.J.; Jaffe, E.K. First structure of full-length mammalian phenylalanine hydroxylase reveals the architecture of an autoinhibited tetramer. Proc. Natl. Acad. Sci. USA 2016, 113, 2394–2399. [Google Scholar] [CrossRef] [Green Version]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Chothia, C. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 1976, 105, 1–12. [Google Scholar] [CrossRef]
- Aguado, C.; Pérez, B.; Ugarte, M.; Desviat, L.R. Analysis of the effect of tetrahydrobiopterin on PAH gene expression in hepatoma cells. FEBS Lett. 2006, 580, 1697–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.; Knappskog, P.M.; Olafsdottir, S.; Døskeland, A.P.; Eiken, H.G.; Svebak, R.M.; Bozzini, M.; Apold, J.; Flatmark, T. Expression of recombinant human phenylalanine hydroxylase as fusion protein in Escherichia coli circumvents proteolytic degradation by host cell proteases. Isolation and characterization of the wild-type enzyme. Biochem. J. 1995, 306 Pt 2, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Goto, N.K.; Williams, K.A.; Deber, C.M. Alpha-helical, but not beta-sheet, propensity of proline is determined by peptide environment. Proc. Natl. Acad. Sci. USA 1996, 93, 6676–6681. [Google Scholar] [CrossRef] [PubMed]
- Blau, N. Genetics of phenylketonuria: Then and now. Hum. Mutat. 2016, 37, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Gámez, A.; Pérez, B.; Ugarte, M.; Desviat, L.R. Expression analysis of phenylketonuria mutations. Effect on folding and stability of the phenylalanine hydroxylase protein. J. Biol. Chem. 2000, 275, 29737–29742. [Google Scholar] [CrossRef] [PubMed]
- Fiege, B.; Blau, N. Assessment of tetrahydrobiopterin (BH4) responsiveness in phenylketonuria. J. Pediatr. 2007, 150, 627–630. [Google Scholar] [CrossRef]
- Wettstein, S.; Underhaug, J.; Perez, B.; Marsden, B.D.; Yue, W.W.; Martinez, A.; Blau, N. Linking genotypes database with locus-specific database and genotype-phenotype correlation in phenylketonuria. Eur J. Hum. Genet. 2015, 302–309. [Google Scholar] [CrossRef]
- Jeannesson-Thivisol, E.; Feillet, F.; Chéry, C.; Perrin, P.; Battaglia-Hsu, S.-F.; Herbeth, B.; Cano, A.; Barth, M.; Fouilhoux, A.; Mention, K.; et al. Genotype-phenotype associations in French patients with phenylketonuria and importance of genotype for full assessment of tetrahydrobiopterin responsiveness. Orphanet J. Rare Dis. 2015, 10, 158. [Google Scholar] [CrossRef]
- Tao, J.; Li, N.; Jia, H.; Liu, Z.; Li, X.; Song, J.; Deng, Y.; Jin, X.; Zhu, J. Correlation between genotype and the tetrahydrobiopterin-responsive phenotype in Chinese patients with phenylketonuria. Pediatr. Res. 2015, 78, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Zurflüh, M.R.; Zschocke, J.; Lindner, M.; Feillet, F.; Chery, C.; Burlina, A.; Stevens, R.C.; Thöny, B.; Blau, N. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum. Mutat. 2008, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Hennermann, J.B.; Roloff, S.; Gebauer, C.; Vetter, B.; von Arnim-Baas, A.; Mönch, E. Long-term treatment with tetrahydrobiopterin in phenylketonuria: Treatment strategies and prediction of long-term responders. Mol. Genet. Metab. 2012, 107, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Jacobs, P.; Fingerhut, R.; Torresani, T.; Thöny, B.; Blau, N.; Baumgartner, M.R.; Rohrbach, M. Positive effect of a simplified diet on blood phenylalanine control in different phenylketonuria variants, characterized by newborn BH4 loading test and PAH analysis. Mol. Genet. Metab. 2012, 106, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Cazzorla, C.; Cegolon, L.; Burlina, A.P.; Celato, A.; Massa, P.; Giordano, L.; Polo, G.; Daniele, A.; Salvatore, F.; Burlina, A.B. Quality of Life (QoL) assessment in a cohort of patients with phenylketonuria. BMC Pub. Health 2014, 14, 1243. [Google Scholar] [CrossRef] [PubMed]
- Scala, I.; Concolino, D.; Della Casa, R.; Nastasi, A.; Ungaro, C.; Paladino, S.; Capaldo, B.; Ruoppolo, M.; Daniele, A.; Bonapace, G.; et al. Long-term follow-up of patients with phenylketonuria treated with tetrahydrobiopterin: A seven years experience. Orphanet J. Rare Dis. 2015, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Ye, J.; Han, L.; Qiu, W.; Zhang, H.; Liang, L.; Gu, X. The predictive value of genetic analyses in the diagnosis of tetrahydrobiopterin (BH4)-Responsiveness in Chinese phenylalanine hydroxylase deficiency patients. Sci. Rep. 2017, 7, 6762. [Google Scholar] [CrossRef] [PubMed]
- Anjema, K.; van Rijn, M.; Hofstede, F.C.; Bosch, A.M.; Hollak, C.E.M.; Rubio-Gozalbo, E.; de Vries, M.C.; Janssen, M.C.H.; Boelen, C.C.A.; Burgerhof, J.G.M.; et al. Tetrahydrobiopterin responsiveness in phenylketonuria: prediction with the 48-hour loading test and genotype. Orphanet J. Rare Dis. 2013, 8, 103. [Google Scholar] [CrossRef]
- Blau, N.; Shen, N.; Carducci, C. Molecular genetics and diagnosis of phenylketonuria: state of the art. Expert Rev. Mol. Diagn. 2014, 14, 655–671. [Google Scholar] [CrossRef]
- Shen, N.; Heintz, C.; Thiel, C.; Okun, J.G.; Hoffmann, G.F.; Blau, N. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol. Genet. Metab. 2016, 117, 328–335. [Google Scholar] [CrossRef]
- Blau, N.; Bélanger-Quintana, A.; Demirkol, M.; Feillet, F.; Giovannini, M.; MacDonald, A.; Trefz, F.K.; van Spronsen, F.J. Optimizing the use of sapropterin (BH4) in the management of phenylketonuria. Mol. Genet. Metab. 2009, 96, 158–163. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, J.J.; Feng, Y.; Zhu, L.L.; Yan, H.; Shi, X.-F.; Chang, A.-M.; Shi, Y.; Wang, P. Mutational spectrum of the phenylalanine hydroxylase gene in patients with phenylketonuria in the central region of China. Scand. J. Clin. Lab. Investig. 2018, 78, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Guldberg, P.; Henriksen, K.F.; Thöny, B.; Blau, N.; Güttler, F. Molecular heterogeneity of nonphenylketonuria hyperphenylalaninemia in 25 Danish patients. Genomics 1994, 21, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Scriver, C.R.; Waters, P.J. Monogenic traits are not simple: Lessons from phenylketonuria. Trends Genet. 1999, 15, 267–272. [Google Scholar] [CrossRef]
- Dipple, K.M.; McCabe, E.R. Modifier genes convert “simple” mendelian disorders to complex traits. Mol. Genet. Metab. 2000, 71, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PAH Mutant | Primer Sequence (5′→3′) | |
|---|---|---|
| p.Asn167Tyr | c.499A > T | CATTGCCTACTACTACCGCC |
| TCAGCAAACTGCTTCCGTC | ||
| p.Thr200Asn | c.599C > A | CTTGTATAAAAACCATGCTTGC |
| GACTTCAGAGTCTTGAACACTGT | ||
| p.Asp229Gly | c.686A > G | CAGCTGGAAGGCGTTTCTCA |
| GGGAATGTTATCTTCATGGAAGCC | ||
| p.Gly239Ala | c.716G > C | ACTTGCACTGCTTTCCGCCT |
| CTGCAGGAACTGAGAAACGTCTTCC | ||
| p.Phe263Ser | c.788T > C | TTCCGAGTCTCCCACTGCACA |
| GGCCAGGCCACCCAAGAAAT | ||
| p.Ala342Pro | c.1024G > C | CTCCATAAAGCCATATGGTGCTG |
| TCTCCTTGTTTGCAGAGCCC | ||
| p.Ile406Met | c.1218A > G | TGCCACAATGCCTCGGCCCT |
| GCAAAGTTCCTTACTTTCTCCTTGGCATCATT | ||
| PAH Variant | Meta-SNP | PredictSNP |
|---|---|---|
| p.Asn167Tyr | Disease | Deleterious |
| 0.542 | 51% | |
| p.Thr200Asn | Disease | Neutral |
| 0.533 | 65% | |
| p.Asp229Gly | Disease | Deleterious |
| 0.542 | 72% | |
| p.Gly239Ala | Disease | Deleterious |
| 0.752 | 87% | |
| p.Phe263Ser | Disease | Deleterious |
| 0.826 | 87% | |
| p.Ala342Pro | Disease | Deleterious |
| 0.824 | 87% | |
| p.Ile406Met | Neutral | Neutral |
| 0.414 | 65% |
| PAH Variant | − GroEL/ES | + GroEL/ES | ||||||
|---|---|---|---|---|---|---|---|---|
| Oligomers (%) | Tetramers (%) | Dimers (%) | Residual Activity (%) | Oligomers (%) | Tetramers (%) | Dimers (%) | Residual Activity (%) | |
| p.Asn167Tyr | 46.6 ± 11.2 | 33.2 ± 5.8 | 20.2 ± 5.8 | 97.8 ± 27.1 | 52.7 ± 2.0 | 38.8 ± 4.2 | 8.5 ± 4.1 | 108.0 ± 27.0 |
| p.Thr200Asn | 49.0 ± 3.5 | 40.5 ± 6.1 | 10.5 ± 6.3 | 92.4 ± 25.9 | 51.7 ± 2.6 | 43.8 ± 3.6 | 4.5 ± 1.2 | 86.3 ± 24.7 |
| p.Asp229Gly | 76.5 ± 13.1 | 4.1 ± 3.7 | 19.3 ± 13.8 | 0 | 79.7 ± 8.4 | 10.2 ± 5.6 | 10.1 ± 2.9 | 0 |
| p.Gly239Ala | 65.9 ± 7.0 | 1.3 ± 2.2 | 32.8 ± 5.8 | 0 | 80.5 ± 8.2 | 5.5 ± 4.8 | 14.0 ± 3.4 | 0.9 ± 1.5 |
| p.Phe263Ser | 67.4 ± 15.4 | 5.5 ± 4.8 | 27.1 ± 10.8 | 0 | 75.4 ± 7.8 | 11.0 ± 4.7 | 13.6 ± 3.7 | 0.9 ± 2.2 |
| p.Ala342Pro | 78.0 ± 11.1 | 0 | 22.0 ± 11.1 | 0 | 81.8 ± 3.9 | 3.8 ± 4.0 | 15.5 ± 7.9 | 9.9 ± 0.7 |
| p.Ile406Met | 50.3 ± 8.9 | 34.2 ± 4.5 | 15.6 ± 4.4 | 83.3 ± 17.5 | 55.5 ± 9.9 | 37.7 ± 7.4 | 6.9 ± 2.3 | 76.7 ± 14.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecimonova, M.; Kluckova, D.; Csicsay, F.; Reblova, K.; Krahulec, J.; Procházkova, D.; Skultety, L.; Kadasi, L.; Soltysova, A. Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase. Genes 2019, 10, 459. https://doi.org/10.3390/genes10060459
Pecimonova M, Kluckova D, Csicsay F, Reblova K, Krahulec J, Procházkova D, Skultety L, Kadasi L, Soltysova A. Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase. Genes. 2019; 10(6):459. https://doi.org/10.3390/genes10060459
Chicago/Turabian StylePecimonova, Martina, Daniela Kluckova, Frantisek Csicsay, Kamila Reblova, Jan Krahulec, Dagmar Procházkova, Ludovit Skultety, Ludevit Kadasi, and Andrea Soltysova. 2019. "Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase" Genes 10, no. 6: 459. https://doi.org/10.3390/genes10060459
APA StylePecimonova, M., Kluckova, D., Csicsay, F., Reblova, K., Krahulec, J., Procházkova, D., Skultety, L., Kadasi, L., & Soltysova, A. (2019). Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase. Genes, 10(6), 459. https://doi.org/10.3390/genes10060459