Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Husbandry
2.2. Lifespan Study and Determining Age at Death
2.3. Female Sexual Maturity
2.4. Plasma IGF-1 Levels
2.5. Statistical Analysis
3. Results
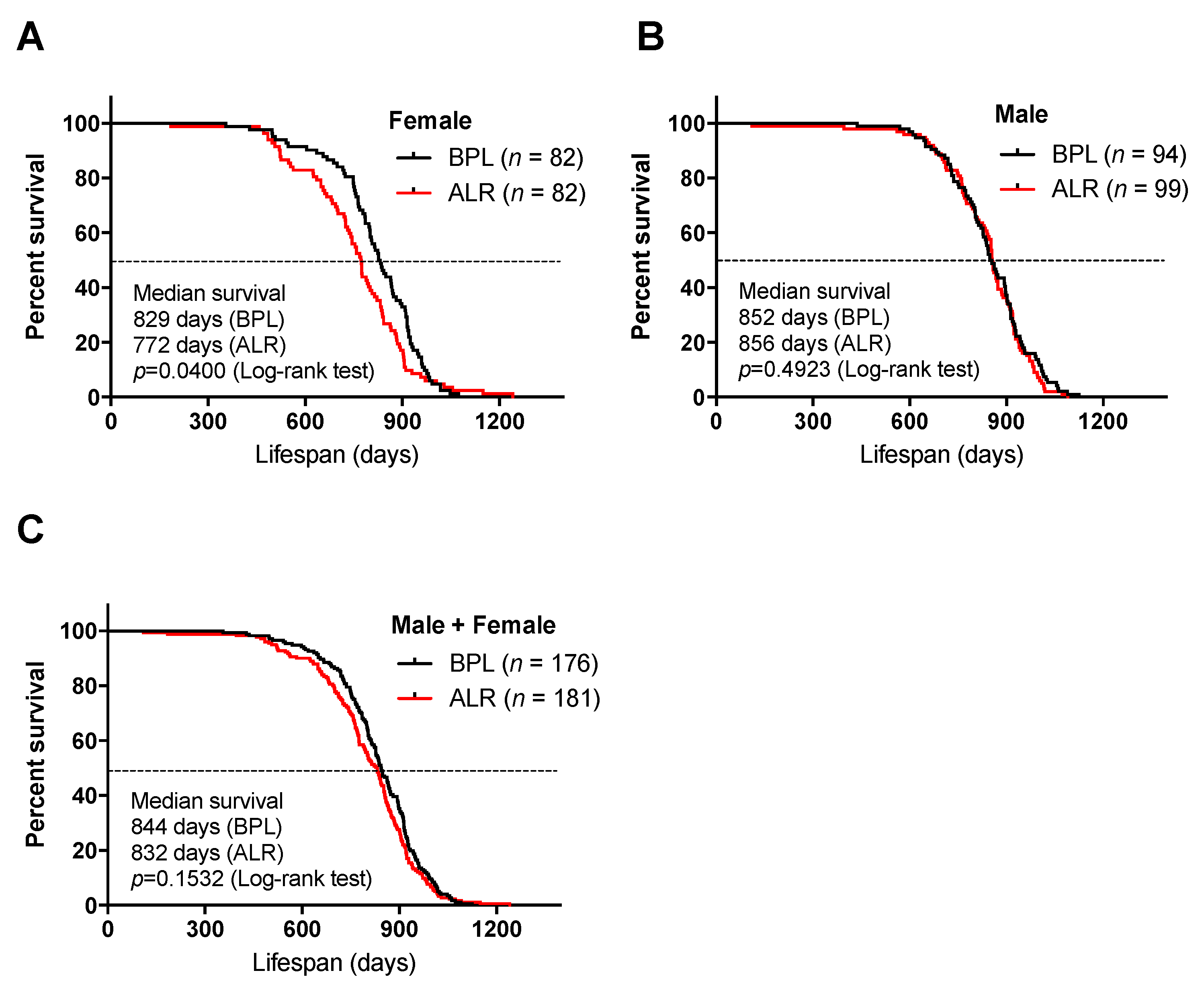
3.1. A Mutation in the mt-Nd2 Gene Results in a Shorter Lifespan in Mice
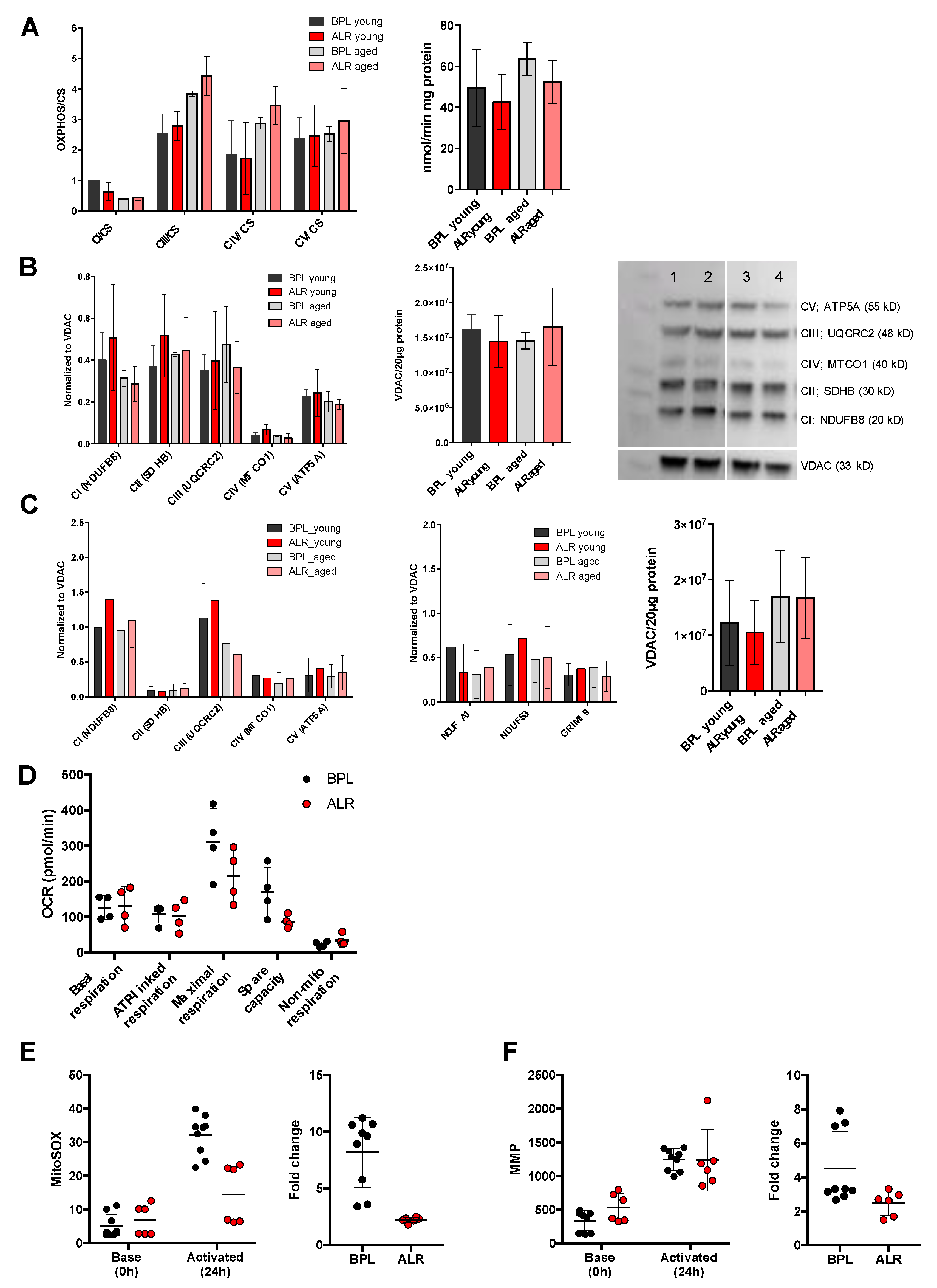
3.2. The mt-Nd2 Mutant Mice Exhibited Mitochondrial Functional Differences under Stress Conditions
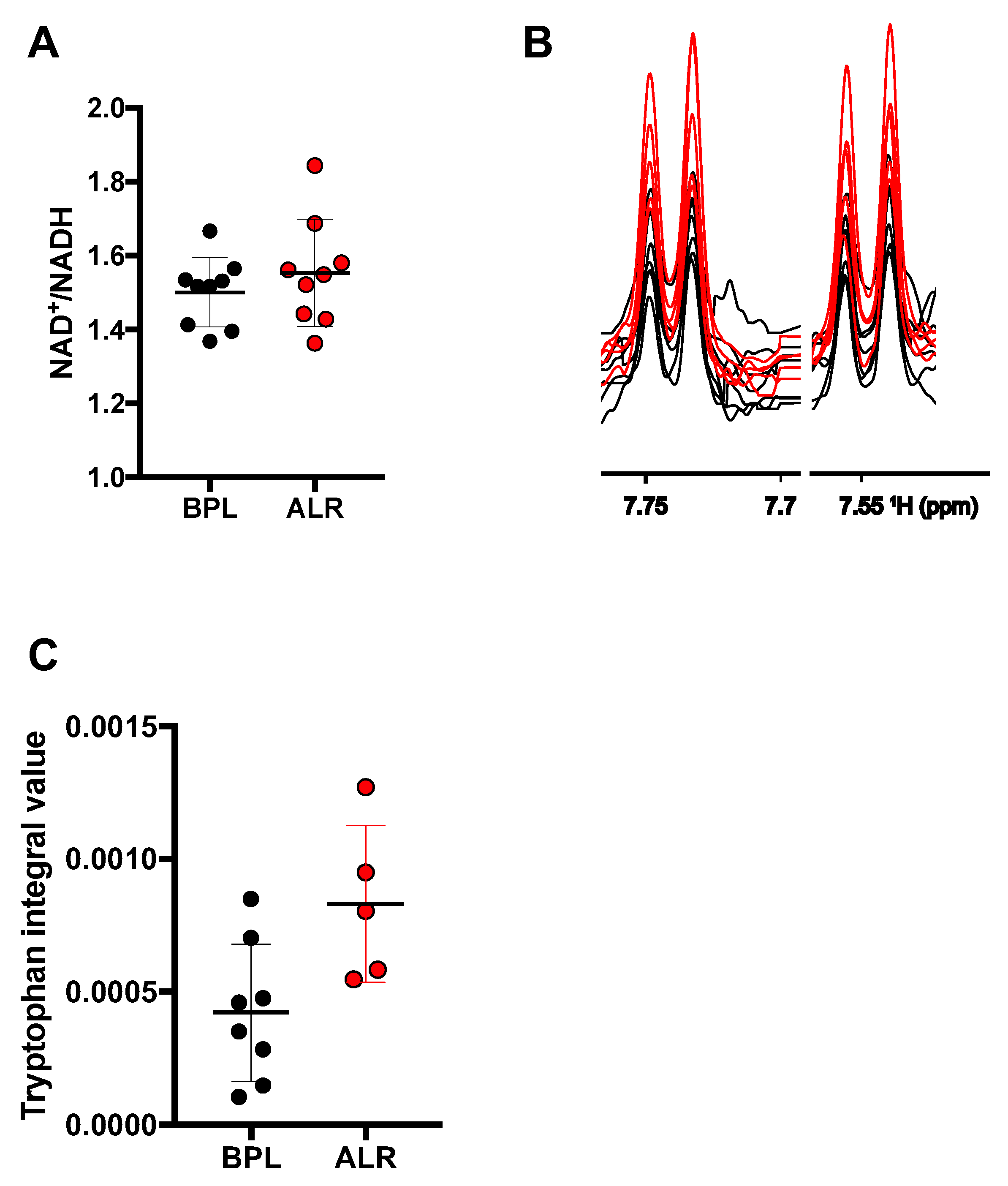
3.3. Higher Levels of Tryptophan Were Observed in the Cells Carrying the mt-Nd2 Mutation
3.4. Ageing-Related Pathways and Mitochondrial Functional Pathways Are Altered in mt-Nd2 Mutant Mice
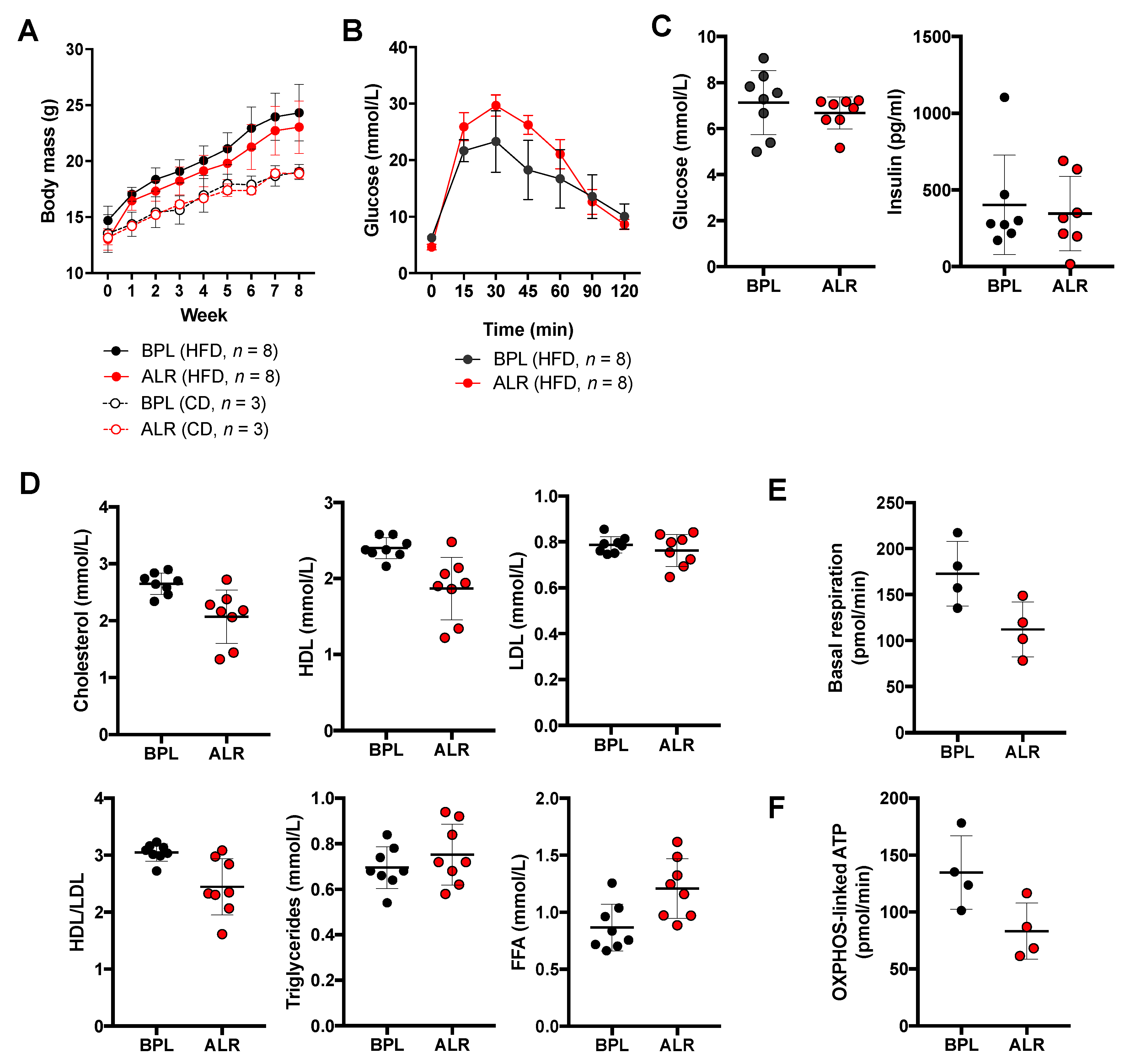
3.5. Earlier Onset of Glucose Intolerance Is Induced by High-Fat Diet Feeding in B6-mtALR Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.J.R.J.; Nijtmans, L.G.; van den Heuvel, L.P.; Smeitink, J.A.M. Mitochondrial complex I:Structure, function and pathology. J. Inherit. Metab. Dis. 2006, 29, 499–515. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Jow, H.; Baty, K.; Johnson, A.; Czapiewski, R.; Saretzki, G.; Treumann, A.; von Zglinicki, T. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 2014, 5, 3837. [Google Scholar] [CrossRef] [Green Version]
- Pujol, C.; Bratic-Hench, I.; Sumakovic, M.; Hench, J.; Mourier, A.; Baumann, L.; Pavlenko, V.; Trifunovic, A. Succinate dehydrogenase upregulation destabilize complex I and limits the lifespan of gas-1 mutant. PLoS ONE 2013, 8, e59493. [Google Scholar] [CrossRef]
- Scialò, F.; Sriram, A.; Fernández-Ayala, D.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Gong, J.S.; Zhang, J.; Yoneda, M.; Yagi, K. Mitochondrial genotype associated with longevity. Lancet 1998, 351, 185–186. [Google Scholar] [CrossRef]
- Ivanova, R.; Lepage, V.; Charron, D.; Schächter, F. Mitochondrial genotype associated with French Caucasian centenarians. Gerontology 1998, 44, 349. [Google Scholar] [CrossRef] [PubMed]
- Guney, O.; Ak, H.; Atay, S.; Ozkaya, A.B.; Aydin, H.H. Mitochondrial DNA polymorphisms associated with longevity in the Turkish population. Mitochondrion 2014, 17, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kokaze, A.; Ishikawa, M.; Matsunaga, N.; Yoshida, M.; Sekine, Y.; Teruya, K.; Takeda, N.; Sumiya, Y.; Uchida, Y.; Takashima, Y. Association of the mitochondrial DNA 5178 A/C polymorphism with serum lipid levels in the Japanese population. Hum. Genet. 2001, 109, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Van der Walt, J.M.; Nicodemus, K.K.; Martin, E.R.; Scott, W.K.; Nance, M.A.; Watts, R.L.; Hubble, J.P.; Haines, J.L.; Koller, W.C.; Lyons, K.; et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003, 72, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, E.; Aida, K.; Chen, J.; Hayashi, J.I.; Isobe, K.; Tawata, M.; Onaya, T. A patient with type 2 diabetes mellitus associated with mutations in calcium sensing receptor gene and mitochondrial DNA. Biochem. Biophys. Res. Commun. 2000, 278, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, A.; Derbeneva, O.; Younes, D.; Keator, D.; Bakken, T.; Lvova, M.; Brandon, M.; Guffanti, G.; Reglodi, D.; Saykin, A.; et al. Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol. Aging 2010, 31, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A.; Hsu, A.-L.; Arantes-Oliveira, N.; Lehrer-Graiwer, J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of Behavior and Aging Specified by Mitochondrial Function During Development. Science 2002, 298, 2398–2401. [Google Scholar] [CrossRef]
- Burman, J.L.; Itsara, L.S.; Kayser, E.-B.; Suthammarak, W.; Wang, A.M.; Kaeberlein, M.; Sedensky, M.M.; Morgan, P.G.; Pallanck, L.J. A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis. Model. Mech. 2014, 7, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Gimsa, U.; Wester-Rosenlöf, L.; Kanitz, E.; Otten, W.; Kunz, M.; Ibrahim, S.M. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 2009, 19, 159–165. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Meng, Q.; Nautiyal, J.; Flurkey, K.; Tsaih, S.-W.; Krier, R.; Parker, M.G.; Harrison, D.E.; Paigen, B. Genetic coregulation of age of female sexual maturation and lifespan through circulating IGF1 among inbred mouse strains. Proc. Natl. Acad. Sci. USA 2012, 109, 8224–8229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Hirose, M.; Schilf, P.; Gupta, Y.; Zarse, K.; Künstner, A.; Fähnrich, A.; Busch, H.; Yin, J.; Wright, M.N.; Ziegler, A.; et al. Low-level mitochondrial heteroplasmy modulates DNA replication, glucose metabolism and lifespan in mice. Sci. Rep. 2018, 8, 5872. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, X.; Lombès, M.; Rha, G.B.; Chi, Y.-I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205. [Google Scholar] [CrossRef] [PubMed]
- El-Assaad, W.; El-Kouhen, K.; Mohammad, A.H.; Yang, J.; Morita, M.; Gamache, I.; Mamer, O.; Avizonis, D.; Hermance, N.; Kersten, S.; et al. Deletion of the gene encoding G0/G 1 switch protein 2 (G0s2) alleviates high-fat-diet-induced weight gain and insulin resistance, and promotes browning of white adipose tissue in mice. Diabetologia 2015, 58, 149–157. [Google Scholar] [CrossRef]
- Ge, X.; Yang, H.; Bednarek, M.A.; Galon-Tilleman, H.; Chen, P.; Chen, M.; Lichtman, J.S.; Wang, Y.; Dalmas, O.; Yin, Y.; et al. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab. 2018, 27, 461–469.e6. [Google Scholar] [CrossRef] [Green Version]
- Päivärinne, H.; Kainulainen, H. DAPIT, a novel protein down-regulated in insulin-sensitive tissues in streptozotocin-induced diabetes. Acta Diabetol. 2001, 38, 83–86. [Google Scholar] [CrossRef]
- Kontro, H.; Hulmi, J.J.; Rahkila, P.; Kainulainen, H. Cellular and tissue expression of DAPIT, a phylogenetically conserved peptide. Eur. J. Histochem. 2012, 56, e18. [Google Scholar] [CrossRef]
- Koc, E.C.; Cimen, H.; Kumcuoglu, B.; Abu, N.; Akpinar, G.; Haque, M.E.; Spremulli, L.L.; Koc, H. Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front. Physiol. 2013, 4, 183. [Google Scholar] [CrossRef] [Green Version]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Benevolenskaya, E.V.; Frolov, M.V. Emerging links between E2F control and mitochondrial function. Cancer Res. 2015, 75, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Mouser, J.; Pitt, J.; Promislow, D.; Kaeberlein, M. Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget 2016, 7, 80131–80139. [Google Scholar] [CrossRef] [PubMed]
- Van der Goot, A.T.; Nollen, E.A.A. Tryptophan metabolism: entering the field of aging and age-related pathologies. Trends Mol. Med. 2013, 19, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Kanamori, H.; Seki, E.; Hoshi, M.; Ohtaki, H.; Yasuda, Y.; Ito, H.; Suetsugu, A.; Nagaki, M.; Moriwaki, H.; et al. L-tryptophan-mediated enhancement of susceptibility to nonalcoholic fatty liver disease is dependent on the mammalian target of rapamycin. J. Biol. Chem. 2011, 286, 34800–34808. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Fang, F.; Chen, J.; Mathews, C.E.; Li, W.; Chu, C.T.; Ding, J.-Q.; Chen, S. Association of the mt-ND2 5178A/C polymorphism with Parkinson’s disease. Neurosci. Lett. 2015, 587, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Künstner, A.; Schilf, P.; Tietjen, A.K.; Jöhren, O.; Huebbe, P.; Rimbach, G.; Rupp, J.; Schwaninger, M.; Busch, H.; et al. A Natural mtDNA Polymorphism in Complex III Is a Modifier of Healthspan in Mice. Int. J. Mol. Sci. 2019, 20, 2359. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirose, M.; Schilf, P.; Zarse, K.; Busch, H.; Fuellen, G.; Jöhren, O.; Köhling, R.; König, I.R.; Richer, B.; Rupp, J.; et al. Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan. Genes 2019, 10, 532. https://doi.org/10.3390/genes10070532
Hirose M, Schilf P, Zarse K, Busch H, Fuellen G, Jöhren O, Köhling R, König IR, Richer B, Rupp J, et al. Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan. Genes. 2019; 10(7):532. https://doi.org/10.3390/genes10070532
Chicago/Turabian StyleHirose, Misa, Paul Schilf, Kim Zarse, Hauke Busch, Georg Fuellen, Olaf Jöhren, Rüdiger Köhling, Inke R. König, Barbara Richer, Jan Rupp, and et al. 2019. "Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan" Genes 10, no. 7: 532. https://doi.org/10.3390/genes10070532
APA StyleHirose, M., Schilf, P., Zarse, K., Busch, H., Fuellen, G., Jöhren, O., Köhling, R., König, I. R., Richer, B., Rupp, J., Schwaninger, M., Seeger, K., Sina, C., Ristow, M., & Ibrahim, S. M. (2019). Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan. Genes, 10(7), 532. https://doi.org/10.3390/genes10070532