Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study

 , , ,
, , ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source and Samples Selection
2.2. RNA-Seq Data Analysis
2.3. Mutational Data Analysis
2.4. sCNA Data Analysis
2.5. Statistical Power and Sample Size Calculation
2.6. Integrated sCNA and RNA-Seq Functional Analysis
3. Results
3.1. Mutational Landscape of TCGA-OV27 Cohort
3.2. Mutational Signatures of TCGA-OV27 Cohort
3.3. Genomic Instability and sCNA Landscape of TCGA-OV27 Cohort
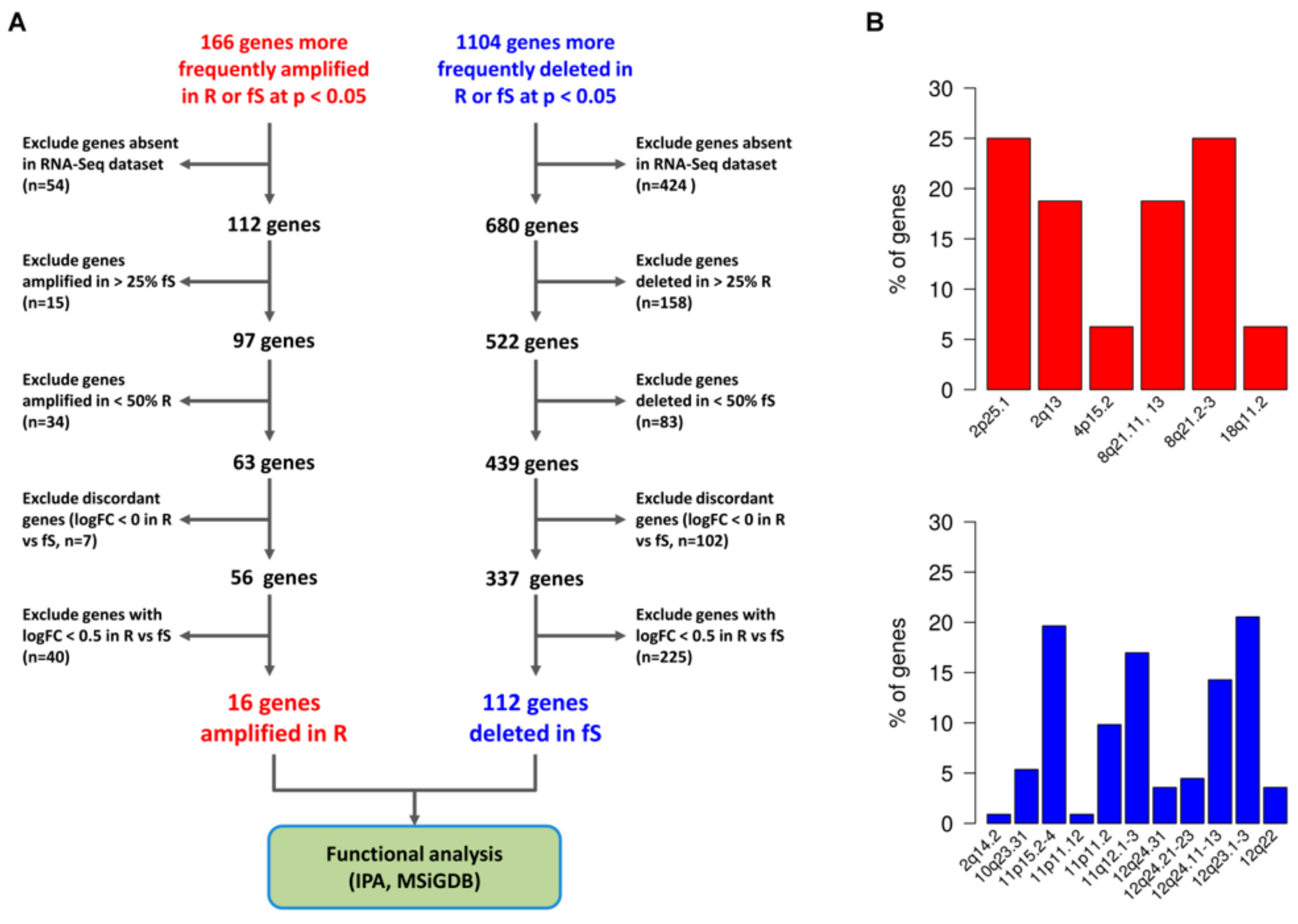
3.4. Association between sCNA and Altered Gene/Pathways Expression in Pt-Sensitivity Classes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 17745–17750. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.C.; Lheureux, S.; Karakasis, K.; Burnier, J.V.; Bruce, J.P.; Clouthier, D.L.; Danesh, A.; Quevedo, R.; Dowar, M.; Hanna, Y.; et al. Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long- and short-term survivors. Genome Med. 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, G.; Goranova, T.E.; De, S.D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Nick, A.M.; Coleman, R.L.; Ramirez, P.T.; Sood, A.K. A framework for a personalized surgical approach to ovarian cancer. Nat. Rev. Clin. Oncol. 2015, 12, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, N.C.; Monteverde, M.; Riba, M.; Lattanzio, L.; Tonissi, F.; Garrone, O.; Heouaine, A.; Gallo, F.; Ceppi, M.; Borghi, F.; et al. Expression profiling and long lasting responses to chemotherapy in metastatic gastric cancer. Int. J. Oncol. 2010, 37, 1219–1228. [Google Scholar] [Green Version]
- Bossi, P.; Bergamini, C.; Siano, M.; Cossu, R.M.; Sponghini, A.P.; Favales, F.; Giannoccaro, M.; Marchesi, E.; Cortelazzi, B.; Perrone, F.; et al. Functional genomics uncover the biology behind the responsiveness of head and neck squamous cell cancer patients to cetuximab. Clin. Cancer Res. 2016, 22, 3961–3970. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Riester, M.; Wei, W.; Waldron, L.; Culhane, A.C.; Trippa, L.; Oliva, E.; Kim, S.H.; Michor, F.; Huttenhower, C.; Parmigiani, G.; et al. Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, S.; Craparotta, I.; Paracchini, L.; Mannarino, L.; Corso, S.; Pezzotta, M.G.; Vescio, M.; Fruscio, R.; Romualdi, C.; Dainese, E.; et al. Multisite analysis of high-grade serous epithelial ovarian cancers identifies genomic regions of focal and recurrent copy number alteration in 3q26.2 and 8q24.3. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Davis, S.; D’Andrea, A.D.; Simpson, K.; Hahn, W.C.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [PubMed]
- Au-Yeung, G.; Lang, F.; Azar, W.J.; Mitchell, C.; Jarman, K.E.; Lackovic, K.; Aziz, D.; Cullinane, C.; Pearson, R.B.; Mileshkin, L.; et al. Selective targeting of Cyclin E1-amplified high-grade serous ovarian cancer by cyclin-dependent kinase 2 and AKT inhibition. Clin. Cancer Res. 2017, 23, 1862–1874. [Google Scholar] [CrossRef]
- Belandia, B.; Powell, S.M.; Garcia-Pedrero, J.M.; Walker, M.M.; Bevan, C.L.; Parker, M.G. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol. Cell Biol. 2005, 25, 1425–1436. [Google Scholar] [CrossRef]
- Fukusumi, T.; Guo, T.W.; Sakai, A.; Ando, M.; Ren, S.; Haft, S.; Liu, C.; Amornphimoltham, P.; Gutkind, J.S.; Califano, J.A. The NOTCH4-HEY1 pathway induces epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Clin. Cancer Res. 2018, 24, 619–633. [Google Scholar] [CrossRef]
- Chen, C.; Wang, X.; Huang, S.; Wang, L.; Han, L.; Yu, S. Prognostic roles of Notch receptor mRNA expression in human ovarian cancer. Oncotarget 2017, 8, 32731–32740. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and epigenomic signatures in ovarian cancer associated with resensitization to platinum drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Gourley, C.; Balmana, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving from poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 27) | R (n = 11) | Fs (n = 16) | |
|---|---|---|---|
| Stage | |||
| III | 23 | 9 | 14 |
| IV | 4 | 2 | 2 |
| Grading | |||
| G2 | 3 | 0 | 3 |
| G3 | 23 | 10 | 13 |
| NA | 1 | 1 | 0 |
| Relapse | |||
| yes | 18 | 11 | 7 |
| no | 9 | 0 | 9 |
| Ingenuity Canonical Pathways | -Log(p-Value) | Genes |
|---|---|---|
| Interferon signaling | 2.87 | OAS1, IFIT1, IFIT3 |
| Oleate biosynthesis II (animals) | 2.8 | FADS1, FADS2 |
| Graft-versus-host Disease signaling | 2.62 | IL1RN, IL36RN, FAS |
| Gαs signaling | 2.4 | CNGB3, GNG3, HCAR3, HCAR2 |
| γ-linolenate biosynthesis II (animals) | 2.33 | FADS1, FADS2 |
| Gαi signaling | 2.15 | APLNR, GNG3, HCAR2, CHRM4 |
| Top Diseases and Functions | Molecules in Network | Score | Focus Molecules |
|---|---|---|---|
| Dermatological diseases and conditions, Organismal injury and abnormalities, immunological disease | ACTA2, ADM, APLNR, Akt, CCKBR, CD6, ERK, ERK1/2, FAS, GLI2, HCAR2, IFIT1, IFIT3, IL1RN, ILK, Interferon alpha, Jnk, MAPK8IP1, Mek, NFkB (complex), OAS1, OAS2, OAS3, P38 MAPK, PI3K (family), PPP1CC, Raf, SH2B3, SLC15A3, SLC43A3, TCR, TRIM22, UBE2L6, UNG, WEE1 | 44 | 24 |
| Gastrointestinal disease, organismal injury and abnormalities, cell death and survival | ADM, AHNAK, ATG7, C3, CFTR, CLDN7, CST5, CTSS, DENND5A, DUSP10, FADS1, FADS2, HAS1, HCAR3, HLA-B, IFNGR1, IL13, IL1B, IL36RN, LBP, MAFF, MS4A4A, NFKBIE, NRIP3, PQLC3, SEL1L3, SLC43A3, SMARCA4, STK33, STMN2, SYT7, TNF, TP63, TUB, WWP1 | 27 | 17 |
| Gene expression, cell cycle, cellular growth and proliferation | ACAD10, BAG1, CBS/CBSL, CCNB2, CDK17, CORO1C, CTR9, DCHS1, ESR1, FGD6, GREB1, HAUS8, HDAC1, HLTF, LTB, NEDD1, NFYB, NR1D1, NR2C1, NR3C1, NUPR1, PLK1, PRMT6, SCUBE2, SMARCE1, SMYD2, SMYD3, SP1, TBX3, TEAD1, TFAP2C, TNFAIP6, TRAFD1, YWHAG, estrogen receptor | 19 | 13 |
| Cellular development, cellular growth and proliferation, cell cycle | CCND1, CDCA2, CDK5, CHD7, CMKLR1, Ctbp, DRAM1, E2F5, ERBB2, GCN1, HCAR1, HEY1, JAG1, LINC-ROR, LIPF, MAFB, MED13L, NOTCH2, NOTCH4, NUAK1, NUMB, OIP5-AS1, PCLAF, PPARGC1A, RFC1, RMST, RUNX3, SLC16A1, SMTN, SOX2, SUV39H1, TMEM119, TMPO, TP53, let-7a-5p | 17 | 12 |
| Gene expression, cell signaling, cellular development | 26s Proteasome, ACACB, ACTA2, AR, ASCL1, ATP6V0D2, BAG1, CD55, CDK5, CDKN1C, CHRM4, CHST1, CKAP4, DAB2, DLL4, FSH, GBP1, H2AFY, HES1, HRK, IER3, LYVE1, Lh, MED12, MTOR, NOTCH1, PGR, PRKD1, PRKD2, SMARCE1, SMTNL1, SSH1, TOP1, TP53I11, YWHAB | 17 | 12 |
| DNA replication, recombination, and repair, cell morphology, cellular function and maintenance | ACTG1, CHPT1, CLOCK, DDX11, DDX5, DTX4, EIF4G2, EP300, FEN1, GATA1, HBB, HNRNPC, HNRNPD, HNRNPU, HUS1, MAX, MYB, OAS3, OTUB1, PARPBP, PCLAF, PCNA, RAD51, RAD9A, RFC1, RHOA, Rnr, SATB1, TMEM241, TP53BP1, TRPV4, USP44, UTP20, XRN2, YBX1 | 15 | 11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugo, M.; Devecchi, A.; De Cecco, L.; Cecchin, E.; Mezzanzanica, D.; Sensi, M.; Bagnoli, M. Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study. Genes 2019, 10, 678. https://doi.org/10.3390/genes10090678
Dugo M, Devecchi A, De Cecco L, Cecchin E, Mezzanzanica D, Sensi M, Bagnoli M. Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study. Genes. 2019; 10(9):678. https://doi.org/10.3390/genes10090678
Chicago/Turabian StyleDugo, Matteo, Andrea Devecchi, Loris De Cecco, Erika Cecchin, Delia Mezzanzanica, Marialuisa Sensi, and Marina Bagnoli. 2019. "Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study" Genes 10, no. 9: 678. https://doi.org/10.3390/genes10090678
APA StyleDugo, M., Devecchi, A., De Cecco, L., Cecchin, E., Mezzanzanica, D., Sensi, M., & Bagnoli, M. (2019). Focal Recurrent Copy Number Alterations Characterize Disease Relapse in High Grade Serous Ovarian Cancer Patients with Good Clinical Prognosis: A Pilot Study. Genes, 10(9), 678. https://doi.org/10.3390/genes10090678