Comparative Study of Pine Reference Genomes Reveals Transposable Element Interconnected Gene Networks

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Datasets
2.2. Extraction of Gene Introns and Flanking Regions
2.3. Analysis of LTR Structure and Transcription Factor Binding Sites (TFBS)
2.4. Gene Networking and Gene Ontology Analysis
3. Results
3.1. Quality of Assembled Genomes and TE Assay
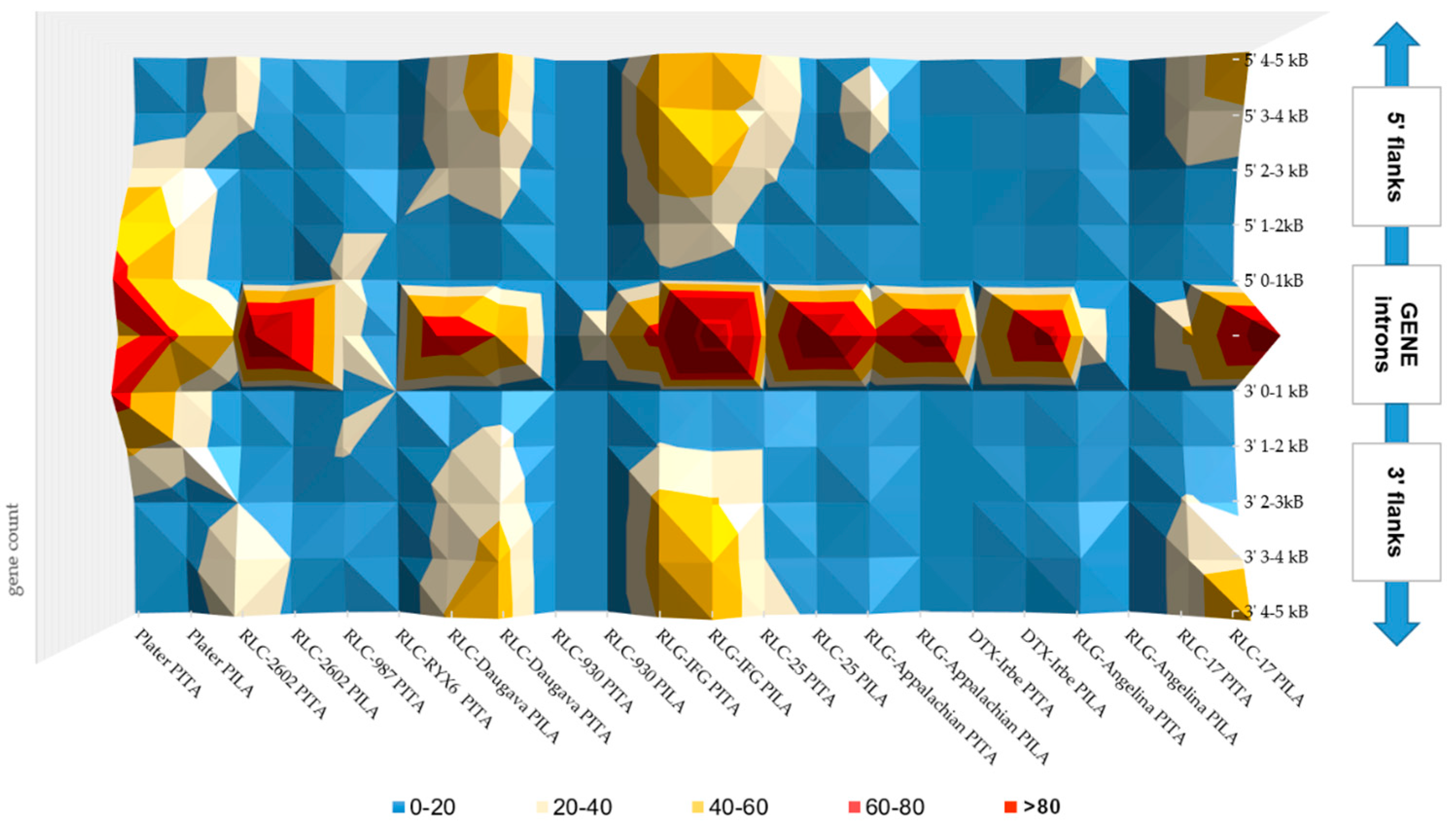
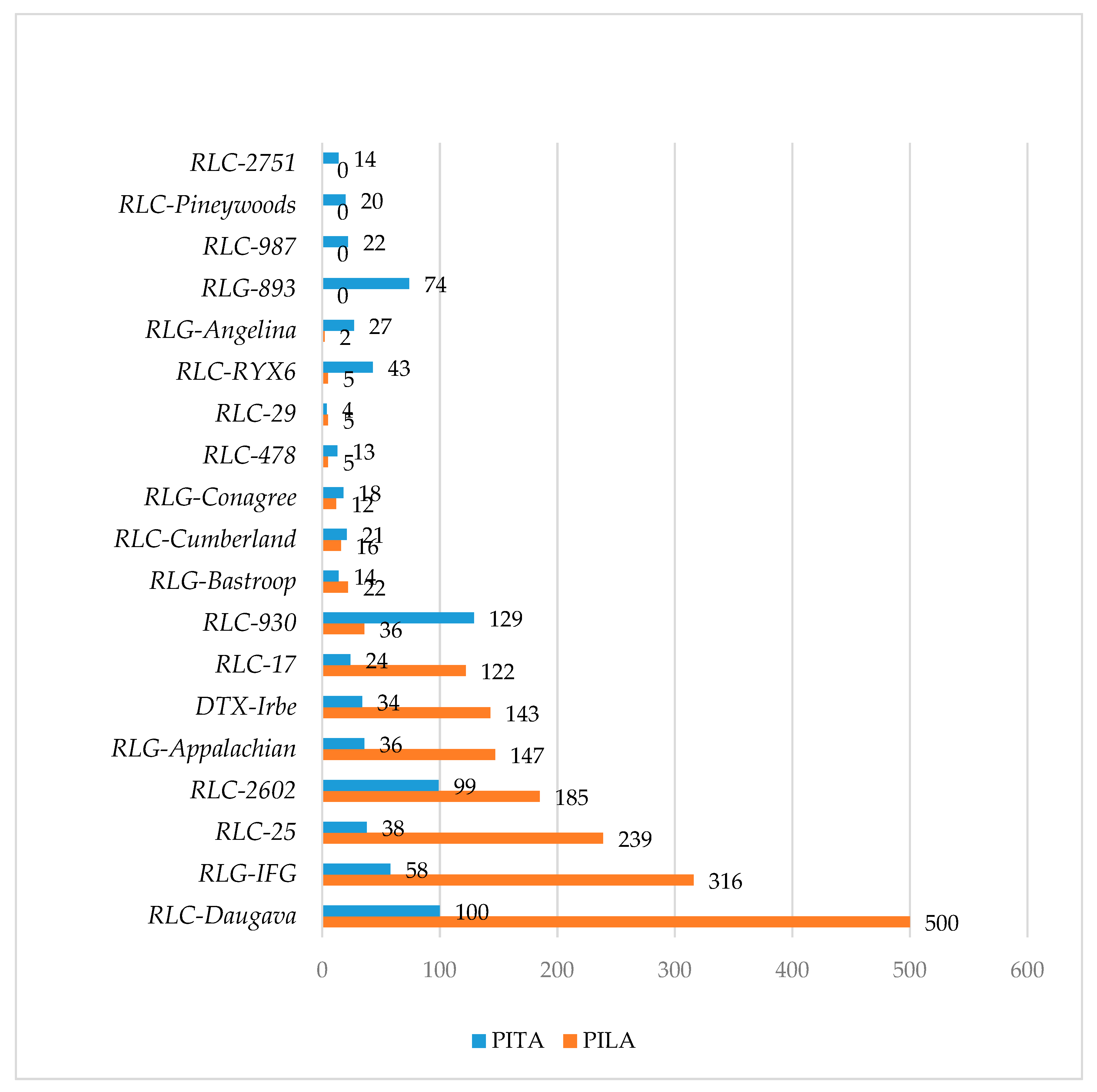
3.2. Topology of TEs in Gene Non-Coding Regions
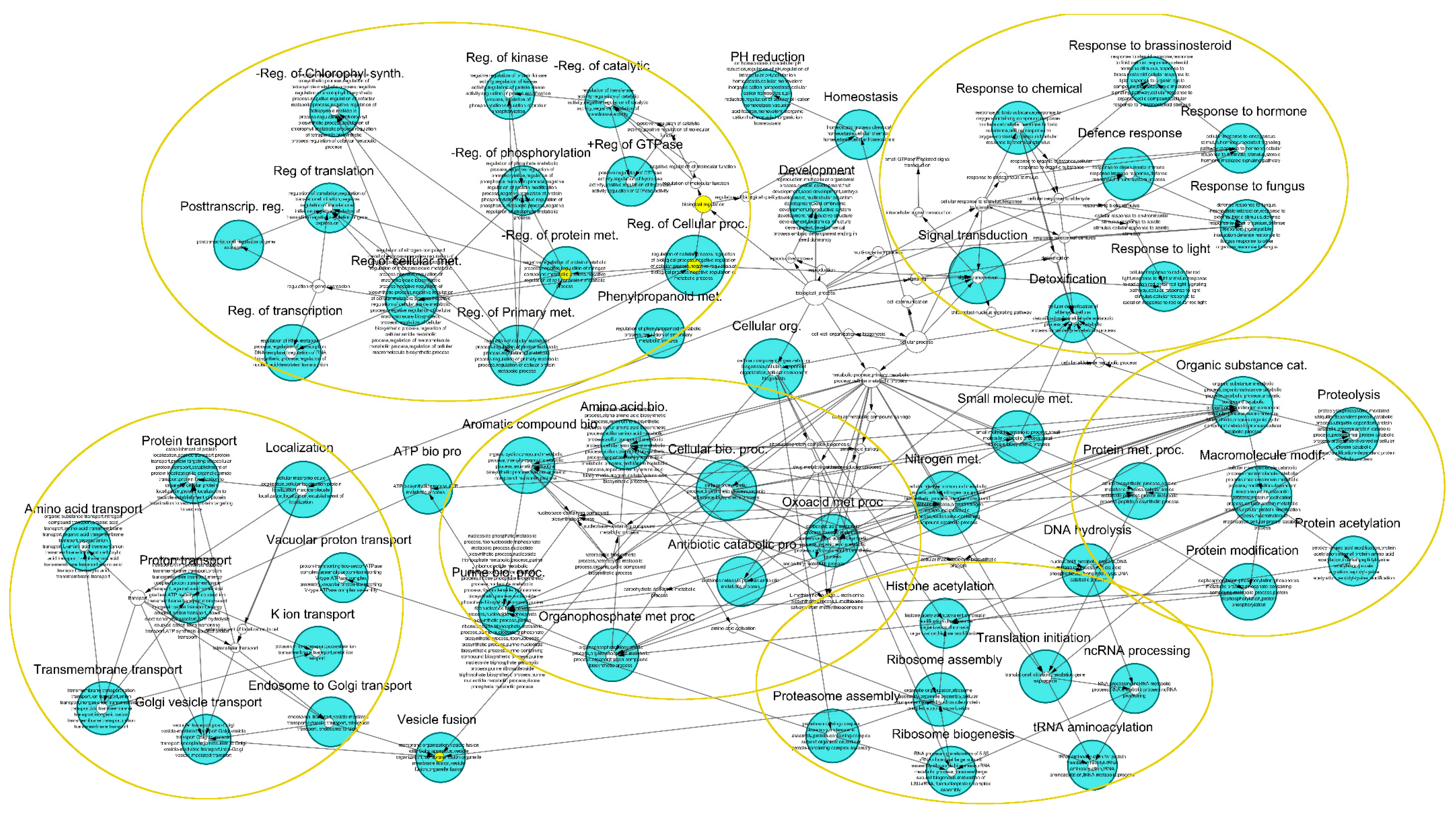
3.3. Gene Ontology Analysis
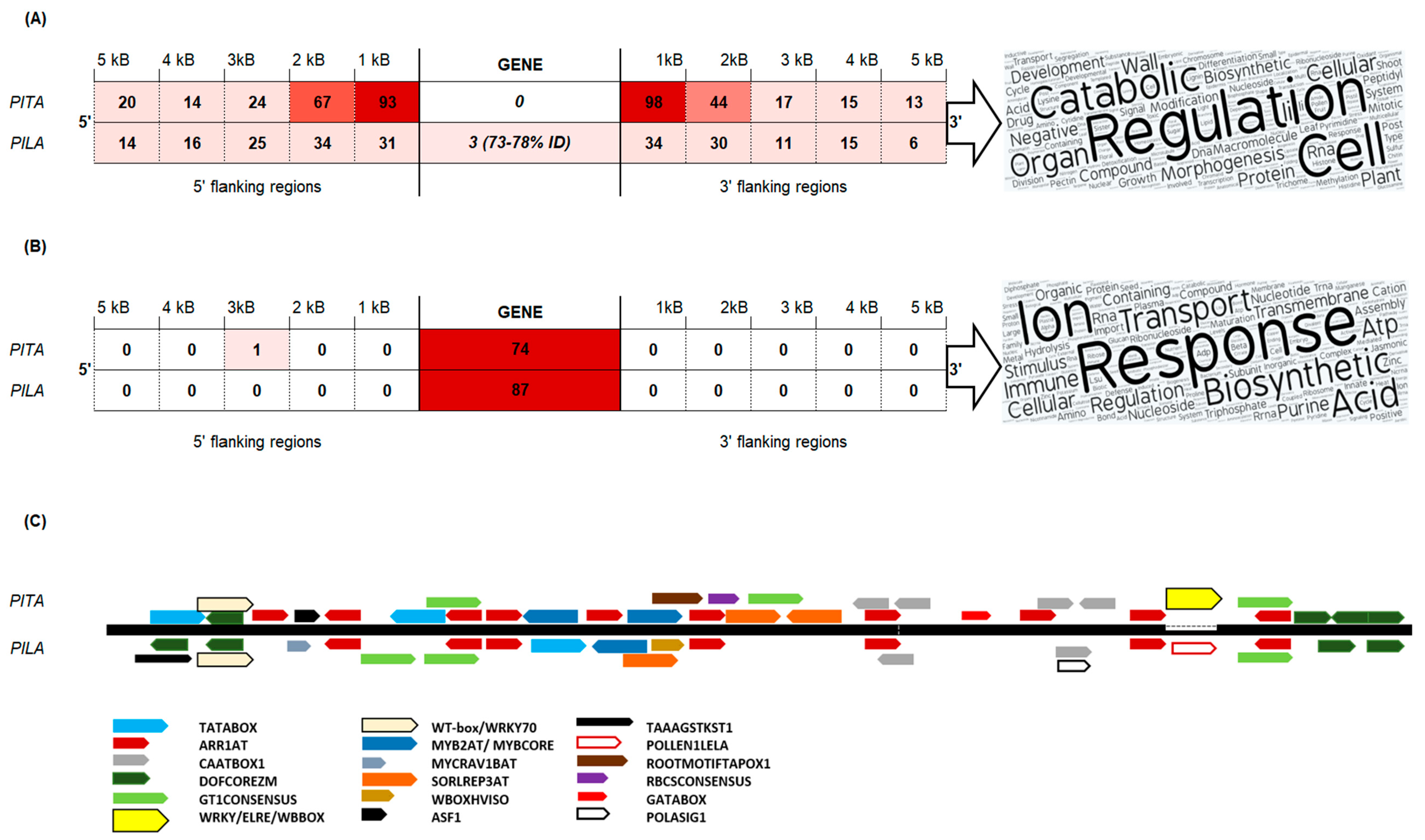
3.4. MITE Plater Distribution in Gene Vicinity and Introns
3.5. DNA Transposon Irbe Forms a Stress-Responsive Gene Network
3.6. Distribution of the Widespread IFG Gypsy RLX
3.7. RLX Daugava Resides in Gene Introns and Flanks
3.8. Genes Appearing in Many TE-Associated Networks
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, H.; Liu, Q.; Li, J.; Jiang, D.; Zhou, L.; Wu, P.; Lu, S.; Li, F.; Zhu, L.; Liu, Z.; et al. Photoperiod-and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res. 2012, 22, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Zhang, Z.; Krause, H.M. Long Noncoding RNAs and Repetitive Elements: Junk or Intimate Evolutionary Partners? Trends Genet. 2019, 35, 892–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessler, S.R. Plant retrotransposons: Turned on by stress. Curr. Biol. 1996, 6, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Capy, P.; Gasperi, G.; Biémont, C.; Bazin, C. Stress and transposable elements: Co-evolution or useful parasites? Heredity (Edinb) 2000, 85, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Tanskanen, J.; Immonen, S.; Nevo, E.; Schulman, A.H. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl. Acad. Sci. USA 2000, 97, 6603–6607. [Google Scholar] [CrossRef] [Green Version]
- Kazazian, H.H. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [Green Version]
- Wendel, J.F.; Greilhuber, J.; Doležel, J.; Leitch, I.J. Plant genome diversity volume 1: Plant genomes, their residents, and their evolutionary dynamics. In Plant Genome Divers Vol 1 Plant Genomes, Their Residents, and Their Evolutionary Dynamics; Springer: Berlin, Germany, 2012; pp. 1–279. [Google Scholar]
- Grandbastien, M.A. LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 403–416. [Google Scholar] [CrossRef]
- Baluska, F.; Gagliano, M.; Witzany, G. Memory and Learning in Plants; Springer: Cham, Germany, 2018. [Google Scholar] [CrossRef]
- Beguiristain, T.; Grandbastien, M.A.; Puigdomènech, P.; Casacuberta, J.M. Three Tnt1 subfamilies show different stress-associated patterns of expression in tobacco. Consequences for retrotransposon control and evolution in plants. Plant Physiol. 2001, 127, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Hadjiargyrou, M.; Delihas, N. The intertwining of transposable elements and non-coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307–13328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Jin, P.; Zhou, X.; Chen, L.; Ma, F. The Role of Transposable Elements in the Origin and Evolution of MicroRNAs in Human. PLoS ONE 2015, 10, e0131365. [Google Scholar] [CrossRef]
- Fray, R.G.; Grierson, D. Identification and genetic analysis of normal and mutant phytoene synthase genes of tomato by sequencing, complementation and co-suppression. Plant Mol. Biol. 1993, 22, 589–602. [Google Scholar] [CrossRef]
- Kashkush, K.; Feldman, M.; Levy, A.A. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 2003, 33, 102–106. [Google Scholar] [CrossRef]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-Induced Mutations in Grape Skin Color. Science 2004, 304, 982. [Google Scholar] [CrossRef]
- Chu, C.-G.; Tan, C.T.; Yu, G.-T.; Zhong, S.; Xu, S.S.; Yan, L. A Novel Retrotransposon Inserted in the Dominant Vrn-B1 Allele Confers Spring Growth Habit in Tetraploid Wheat (Triticum turgidum L.). G3 (Bethesda) 2011, 1, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons Control Fruit-Specific, Cold-Dependent Accumulation of Anthocyanins in Blood Oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, T.; Eulgem, T. An alternative polyadenylation mechanism coopted to the Arabidopsis RPP7 gene through intronic retrotransposon domestication. Proc. Natl. Acad. Sci. USA 2013, 110, E3535–E3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Cuzick, A.; Lu, X.M.; Wang, J.; Katiyar, N.; Tsuchiya, T.; Le Roch, K.; McDowell, J.M.; Holub, E.; Eulgem, T. The Arabidopsis RRM domain protein EDM 3 mediates race-specific disease resistance by controlling H3K9me2-dependent alternative polyadenylation of RPP 7 immune receptor transcripts. Plant J. 2019, 97, 646–660. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Sugimoto, K.; Otsuki, H.; Hirochika, H. A 13-bp cis-regulatory element in the LTR promoter of the tobacco retrotransposon Tto1 is involved in responsiveness to tissue culture, wounding, methyl jasmonate and fungal elicitors. Plant J. 1999, 18, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Varagona, M.J.; Purugganan, M.; Wessler, S.R. Alternative splicing induced by insertion of retrotransposons into the maize waxy gene. Plant Cell 1992, 4, 811–820. [Google Scholar]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; van der Knaap, E. A Retrotransposon-Mediated Gene Duplication Underlies Morphological Variation of Tomato Fruit. Science 2008, 319, 1527–1530. [Google Scholar] [CrossRef]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable Elements: An Abundant and Natural Source of Regulatory Sequences for Host Genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef]
- Mita, P.; Boeke, J.D. How retrotransposons shape genome regulation. Curr. Opin. Genet. Dev. 2016, 37, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant Retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Jimenez-Lopez, J.C.; Iwata, A.; Gill, N.; Jackson, S.A. Functional and Structural Divergence of an Unusual LTR Retrotransposon Family in Plants. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasu, K.; Schulman, A.H.; Lahaye, T.; Schulze-Lefert, P. A contiguous 66-kb barley DNA sequence provides evidence for reversible genome expansion. Genome Res. 2000, 10, 908–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalendar, R.; Vicient, C.M.; Peleg, O.; Anamthawat-Jonsson, K.; Bolshoy, A.; Schulman, A.H. Large Retrotransposon Derivatives: Abundant, Conserved but Nonautonomous Retroelements of Barley and Related Genomes. Genetics 2004, 166, 1437–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Martinez, J.; Martínez-Izquierdo, J.A. Study on the Evolution of the Grande Retrotransposon in the Zea Genus. Mol. Biol. Evol. 2003, 20, 831–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macas, J.; Neumann, P. Ogre elements—A distinct group of plant Ty3/gypsy-like retrotransposons. Gene 2007, 390, 108–116. [Google Scholar] [CrossRef]
- Rho, M.; Choi, J.-H.H.; Kim, S.; Lynch, M.; Tang, H. De novo identification of LTR retrotransposons in eukaryotic genomes. BMC Genom. 2007, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- You, F.M.; Cloutier, S.; Shan, Y.; Ragupathy, R. LTR Annotator: Automated Identification and Annotation of LTR Retrotransposons in Plant Genomes. Int. J. Biosci. Biochem. Bioinform. 2015, 5, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Brierley, C.; Flavell, A.J. The retrotransposon Copia controls the relative levels of its gene products post-transcriptionally be differential expression from its two major mRNAs. Nucleic Acids Res. 1990, 18, 2947–2951. [Google Scholar] [CrossRef] [Green Version]
- Schulman AH (2013) Retrotransposon replication in plants. Curr. Opin. Virol. 1990, 3, 604–614.
- Kalendar, R.; Raskina, O.; Belyayev, A.; Schulman, A.H. Long tandem arrays of Cassandra retroelements and their role in genome dynamics in plants. Int. J. Mol. Sci. 2020, 21, 2931. [Google Scholar] [CrossRef]
- Sabot, F.; Schulman, A.H. Template switching can create complex LTR retrotransposon insertions in Triticeae genomes. BMC Genom. 2007, 8, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yang, F.; Schulman, A.H.; Zhu, J.; Jia, Y.; Wang, J.; Zhang, X.-Q.; Jia, Q.; Hua, W.; Yang, J.; et al. Gene Deletion in Barley Mediated by LTR-retrotransposon BARE. Sci. Rep. 2017, 7, 43766. [Google Scholar] [CrossRef] [Green Version]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.-K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; et al. Nested Retrotransposons in the Intergenic Regions of the Maize Genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Bureau, T.E.; Wessler, S.R. Stowaway: A new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell 1994, 6, 907–916. [Google Scholar] [PubMed] [Green Version]
- Wessler, S.R.; Bureau, T.E.; White, S.E. LTR-retrotransposons and MITEs: Important players in the evolution of plant genomes. Curr. Opin. Genet. Dev. 1995, 5, 814–821. [Google Scholar] [CrossRef]
- Casa, A.M.; Brouwer, C.; Nagel, A.; Wang, L.; Zhang, Q.; Kresovich, S.; Wessler, S.R. The MITE family Heartbreaker (Hbr): Molecular markers in maize. Proc. Natl. Acad. Sci. USA 2000, 97, 10083–10089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purugganan, M.D.; Wessler, S.R. Transposon signatures: Species-specific molecular markers that utilize a class of multiple-copy nuclear DNA. Mol. Ecol. 1995, 4, 265–270. [Google Scholar] [CrossRef]
- Waugh, R.; McLean, K.; Flavell, A.J.; Pearce, S.R.; Kumar, A.; Thomas, B.B.T.; Powell, W. Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP). Mol. Genet. Genom. 1997, 253, 687–694. [Google Scholar] [CrossRef]
- Flavell, A.J.; Knox, M.R.; Pearce, S.R.; Ellis, T.H.N. Retrotransposon-based insertion polymorphisms (RBIP) for high throughput marker analysis. Plant J. 1998, 16, 643–650. [Google Scholar] [CrossRef]
- Kentner, E.K.; Arnold, M.L.; Wessler, S.R. Characterization of high-copy-number retrotransposons from the large genomes of the Louisiana iris species and their use as molecular markers. Genetics 2003, 164, 685–697. [Google Scholar] [PubMed]
- Kalendar, R.; Schulman, A.H. IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat. Protoc. 2006, 1, 2478–2484. [Google Scholar] [CrossRef]
- Kalendar, R.; Flavell, A.J.; Ellis, T.H.N.; Sjakste, T.; Moisy, C.; Schulman, A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity (Edinb) 2011, 106, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Kalendar, R.; Amenov, A.; Daniyarov, A. Use of retrotransposon-derived genetic markers to analyse genomic variability in plants. Funct. Plant Biol. 2019, 46, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Monden, Y.; Tahara, M. Plant Transposable Elements and Their Application to Genetic Analysis via High-throughput Sequencing Platform. Hortic. J. 2015, 84, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh; Nandini, B. Miniature inverted-repeat transposable elements (MITEs), derived insertional polymorphism as a tool of marker systems for molecular plant breeding. Mol. Biol. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Morse, A.M.; Peterson, D.G.; Islam-Faridi, M.N.; Smith, K.E.; Magbanua, Z.; Garcia, S.A.; Kubisiak, T.L.; Amerson, H.V.; Carlson, J.E.; Nelson, C.D.; et al. Evolution of genome size and complexity in Pinus. PLoS ONE 2009, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kovach, A.; Wegrzyn, J.L.; Parra, G.; Holt, C.; Bruening, G.E.; Loopstra, C.A.; Hartigan, J.; Yandell, M.; Langley, C.H.; Korf, I.; et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genom. 2010, 11, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; De Jong, P.J.; et al. Sequencing and assembly of the 22-gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E.; et al. Sequence of the Sugar Pine Megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.W.; Puiu, D.; Wegrzyn, J.L.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. Gigascience 2017, 6, 1–4. [Google Scholar]
- Crepeau, M.W.; Langley, C.H.; Stevens, K.A. From Pine Cones to Read Clouds: Rescaffolding the Megagenome of Sugar Pine (Pinus lambertiana). G3 2017, 7, 1563–1568. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.-S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martínez-García, P.J.; et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef] [Green Version]
- Kossack, D.S.; Kinlaw, C.S. IFG, a gypsy-like retrotransposon in Pinus (Pinaceae), has an extensive history in pines. Plant Mol. Biol. 1999, 39, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Voronova, A.; Belevich, V.; Korica, A.; Rungis, D. Retrotransposon distribution and copy number variation in gymnosperm genomes. Tree Genet. Genom. 2017. [Google Scholar] [CrossRef]
- Voronova, A.; Belevich, V.; Jansons, A.; Rungis, D. Stress-induced transcriptional activation of retrotransposon-like sequences in the Scots pine (Pinus sylvestris L.) genome. Tree Genet Genom. 2014, 10, 937–951. [Google Scholar] [CrossRef]
- Voronova, A. Retrotransposon expression in response to in vitro inoculation with two fungal pathogens of Scots pine (Pinus sylvestris L.). BMC Res. Notes 2019, 12, 243. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-performance genomic feature operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betley, J.N.; Frith, M.C.; Graber, J.H.; Choo, S.; Deshler, J.O. A ubiquitous and conserved signal for RNA localization in chordates. Curr. Biol. 2002, 12, 1756–1761. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Solovyev, V.V.; Shahmuradov, I.A.; Salamov, A.A. Identification of Promoter Regions and Regulatory Sites. Methods Mol. Biol. 2010, 674, 57–83. [Google Scholar]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008. [Google Scholar] [CrossRef] [Green Version]
- Van Bel, M.; Proost, S.; Wischnitzki, E.; Movahedi, S.; Scheerlinck, C.; Van de Peer, Y.; Vandepoele, K. Dissecting Plant Genomes with the PLAZA Comparative Genomics Platform. Plant Physiol. 2012, 158, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goenawan, I.H.; Bryan, K.; Lynn, D.J. DyNet: Visualization and analysis of dynamic molecular interaction networks. Bioinformatics 2016, 32, 2713–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Miguel, C.; Simões, M.; Oliveira, M.M.; Rocheta, M. Envelope-like retrotransposons in the plant kingdom: Evidence of their presence in gymnosperms (Pinus pinaster). J. Mol. Evol. 2008, 67, 517–525. [Google Scholar]
- Candar-Cakir, B.; Arican, E.; Zhang, B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific microRNAs and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Yu, H.; Zhao, G.; Huang, Q.; Lu, Y.; Ouyang, B. Profiling of drought-responsive microRNA and mRNA in tomato using high-throughput sequencing. BMC Genom. 2017, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ibeas, D.; Martinez-Garcia, P.J.; Famula, R.A.; Delfino-Mix, A.; Stevens, K.A.; Loopstra, C.A.; Langley, C.H.; Neale, D.B.; Wegrzyn, J.L. Assessing the Gene Content of the Megagenome: Sugar Pine (Pinus lambertiana). G3 2016, 6, 3787–3802. [Google Scholar] [CrossRef] [Green Version]
- Perera, D.; Magbanua, Z.V.; Thummasuwan, S.; Mukherjee, D.; ArickII, M.; Chouvarine, P.; Nairn, C.J.; Schmutz, J.; Jenkins, J.; Dean, J.F.D.; et al. Exploring the loblolly pine (Pinus taeda L.) genome by BAC sequencing and Cot analysis. Gene 2018, 663, 165–177. [Google Scholar] [CrossRef]
- Huntley, R.P.; Binns, D.; Dimmer, E.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A user tutorial for the web-based Gene Ontology browser. Database 2009, 2009, bap010. [Google Scholar] [CrossRef]
- Bergman, C.M.; Quesneville, H. Discovering and detecting transposable elements in genome sequences. Brief. Bioinform. 2007, 8, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.; Promponas, V.; et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Claros, M.G.; Bautista, R.; Guerrero-Fernández, D.; Benzerki, H.; Seoane, P.; Fernández-Pozo, N. Why assembling plant genome sequences is so challenging. Biology (Basel) 2012, 1, 439–459. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes (Basel) 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, R.; Zhao, Y.; Zhang, H.; Fan, G.; Liu, X.; Zhou, W.; Shi, C.; Wang, J.; Liu, W.; Liang, X.; et al. Draft genome of the living fossil Ginkgo biloba. Gigascience 2016, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Inagaki, Y.; Satoh, H.; Hoshino, A.; Iida, S. Capture of a genomic HMG domain sequence by the En/Spm -related transposable element Tpn1 in the Japanese morning glory. Mol. Genet. Genom. 1999, 261, 447–451. [Google Scholar] [CrossRef]
- Zabala, G.; Vodkin, L.O. The wp mutation of Glycine max carries a gene-fragment-rich transposon of the CACTA superfamily. Plant Cell 2005, 17, 2619–2632. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Stein, J.C.; Liang, C.; Zhang, J.; Fulton, R.S.; Baucom, R.S.; De Paoli, E.; Zhou, S.; Yang, L.; Han, Y.; et al. Detailed analysis of a contiguous 22-Mb region of the maize genome. PLoS Genet. 2009, 5, e1000728. [Google Scholar] [CrossRef] [Green Version]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Casacuberta, J.M.; Grandbastien, M.A. Characterisation of LTR sequences involved in the protoplast specific expression of the tobacco Tnt1 retrotransposon. Nucleic Acids Res. 1993, 21, 2087–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabot, F.; Sourdille, P.; Bernard, M. Advent of a new retrotransposon structure: The long form of the Veju elements. Genetica 2005, 125, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Vicient, C.M.; Kalendar, R.; Schulman, A.H. Variability, recombination, and mosaic evolution of the barley BARE-1 retrotransposon. J. Mol. Evol. 2005, 61, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Liu, J.; Xu, Y.; Liu, X.; Zhang, S.; Ma, J.; Du, J. TARE1, a Mutated Copia-Like LTR Retrotransposon Followed by Recent Massive Amplification in Tomato. PLoS ONE 2013. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V.R.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Maier, F.; Zwicker, S.; Hückelhoven, A.; Meissner, M.; Funk, J.; Pfitzner, A.J.P.; Pfitzner, U.M. Nonexpressor of Pathogenesis-Related Proteins1 (NPR1) and some NPR1-related proteins are sensitive to salicylic acid. Mol. Plant Pathol. 2011, 12, 73–91. [Google Scholar] [CrossRef]
- Yanagisawa, S. Dof Domain Proteins: Plant-Specific Transcription Factors Associated with Diverse Phenomena Unique to Plants. Plant Cell Physiol. 2004, 45, 386–391. [Google Scholar] [CrossRef] [Green Version]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Sasaki, N.; Tsuge, T.; Aoyama, T.; Oka, A. ARR1 Directly Activates Cytokinin Response Genes that Encode Proteins with Diverse Regulatory Functions. Plant Cell Physiol. 2007, 48, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Honma, T.; Takashi, A.; Sato, S.; Kato, T.; Tabata, S.; Oka, A. ARR1, a transcription factor for genes immediately responsive to cytokinins. Science 2001, 294, 1519–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, M.C.; Martin, R.C.; Mok, D.W.S. Cytokinins: Biosynthesis, metabolism and perception. Vitr. Cell. Dev. Biol.-Plant 2000, 36, 102–107. [Google Scholar] [CrossRef]
- Hwang, I.; Sheen, J.; Müller, B. Cytokinin Signaling Networks. Annu. Rev. Plant Biol. 2012, 63, 353–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, M.C. Cytokinin and Plant Development—An Overview. In Cytokinins Chemistry, Activity, and Function; Mok, D.W.S., Mok, M.C., Eds.; CRC Press: Ann Arbor, MI, USA, 1994; pp. 155–166. [Google Scholar]
- Argueso, C.T.; Ferreira, F.J.; Epple, P.; To, J.P.C.; Hutchison, C.E.; Schaller, G.E.; Dangl, J.L.; Kieber, J.J. Two-component elements mediate interactions between cytokinin and salicylic acid in plant immunity. PLoS Genet. 2012. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yin, J.; Xiao, M.; Mason, A.S.; Gao, C.; Liu, H.; Li, J.; Fu, D. Characterization of Structure, Divergence and Regulation Patterns of Plant Promoters. J. Mol. Biol. Res. 2013, 3, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.K.; Dalal, V.; Singh, A.K.; Singh, N.K.; Mohapatra, T. Genic non-coding microsatellites in the rice genome: Characterization, marker design and use in assessing genetic and evolutionary relationships among domesticated groups. BMC Genom. 2009, 10, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krom, N.; Recla, J.; Ramakrishna, W. Analysis of genes associated with retrotransposons in the rice genome. Genetica 2008, 134, 297–310. [Google Scholar] [CrossRef]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable Elements Contribute to Activation of Maize Genes in Response to Abiotic Stress. PLoS Genet. 2015. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhang, W.; Chen, L.; Wang, L.; Marand, A.P.; Wu, Y.; Jiang, J. Proliferation of regulatory DNA elements derived from transposable elements in the maize genome. Plant Physiol. 2018, 176, 2789–2803. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.Y.; Vorst, O.; Fiers, M.W.E.J.; Stiekema, W.J.; Nap, J.P. In plants, highly expressed genes are the least compact. Trends Genet. 2006, 22, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Colinas, J.; Schmidler, S.C.; Bohrer, G.; Iordanov, B.; Benfey, P.N. Intergenic and genic sequence lengths have opposite relationships with respect to gene expression. PLoS ONE 2008. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, S.; Rau, D.; Porceddu, A. Mutational biases and selective forces shaping the structure of Arabidopsis genes. PLoS ONE 2009. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Bansal, M. Variation of gene expression in plants is influenced by gene architecture and structural properties of promoters. PLoS ONE 2019. [Google Scholar] [CrossRef]
- Mizuno, M.; Kanehisa, M. Distribution profiles of GC content around the translation initiation site in different species. FEBS Lett. 1994, 352, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Ming, R.; Yu, Q. Comparative Analysis of GC Content Variations in Plant Genomes. Trop. Plant Biol. 2016, 9, 136–149. [Google Scholar] [CrossRef]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, S.; Ast, G. When epigenetics meets alternative splicing: The roles of DNA methylation and GC architecture. Epigenomics 2013, 5, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.N.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Singer, T.; McConnell, M.J.; Marchetto, M.C.N.; Coufal, N.G.; Gage, F.H. LINE-1 retrotransposons: Mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010, 33, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.R.; Gill, G.P.; Kuntz, R.J.; Langley, C.H.; Neale, D.B. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 2004, 101, 15255–15260. [Google Scholar] [CrossRef] [Green Version]
- Casacuberta, J.M.; Santiago, N. Plant LTR-retrotransposons and MITEs: Control of transposition and impact on the evolution of plant genes and genomes. Gene 2003, 311, 1–11. [Google Scholar] [CrossRef]
- Liu, B.; Shan, X.H.; Liu, Z.L.; Dong, Z.Y.; Wang, Y.M.; Chen, Y.; Lin, X.Y.; Long, L.K.; Han, F.P.; Dong, Y.S. Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (Zizania latifolia Griseb.). Mol. Biol. Evol. 2005, 22, 976–990. [Google Scholar]
- Lu, C.; Chen, J.; Zhang, Y.; Hu, Q.; Su, W.; Kuang, H. Miniature Inverted-Repeat Transposable Elements (MITEs) Have Been Accumulated through Amplification Bursts and Play Important Roles in Gene Expression and Species Diversity in Oryza sativa. Mol. Biol. Evol. 2012, 29, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- To, T.K.; Saze, H.; Kakutani, T. DNA Methylation within Transcribed Regions. Plant Physiol. 2015, 168, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Nandha, P.S.; Singh, J. Transposon-based genetic diversity assessment in wild and cultivated barley. Crop. J. 2017, 5, 296–304. [Google Scholar] [CrossRef]
- Stelmach, K.; Macko-Podgórni, A.; Machaj, G.; Grzebelus, D. Miniature Inverted Repeat Transposable Element Insertions Provide a Source of Intron Length Polymorphism Markers in the Carrot (Daucus carota L.). Front. Plant Sci. 2017, 8, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurka, J.; Bao, W.; Kojima, K.K. Families of transposable elements, population structure and the origin of species. Biol. Direct 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrato-Capuchina, A.; Matute, D.R. The Role of Transposable Elements in Speciation. Genes (Basel) 2018, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Friesen, N.; Brandes, A.; Heslop-Harrison, J.S. Diversity, origin, and distribution of retrotransposons (gypsy and copia) in conifers. Mol. Biol. Evol. 2001, 18, 1176–1188. [Google Scholar] [CrossRef]
- Stone, J.M.; Walker, J.C. Plant protein kinase families and signal transduction. Plant Physiol. 1995, 108, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehti-Shiu, M.D.; Zou, C.; Hanada, K.; Shiu, S.-H. Evolutionary history and stress regulation of plant receptor-like kinase/pelle genes. Plant Physiol. 2009, 150, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehti-Shiu, M.D.; Shiu, S.-H. Diversity, classification and function of the plant protein kinase superfamily. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2619–2639. [Google Scholar] [CrossRef] [Green Version]
- White-Gloria, C.; Johnson, J.J.; Marritt, K.; Kataya, A.; Vahab, A.; Moorhead, G.B. Protein Kinases and Phosphatases of the Plastid and Their Potential Role in Starch Metabolism. Front. Plant Sci. 2018, 9, 1032. [Google Scholar] [CrossRef]
- West, P.T.; Li, Q.; Ji, L.; Eichten, S.R.; Song, J.; Vaughn, M.W.; Schmitz, R.J.; Springer, N.M. Genomic Distribution of H3K9me2 and DNA Methylation in a Maize Genome. PLoS ONE 2014, 9, e105267. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Miyazaki, Y.; Takuno, S.; Saze, H. Epigenetic regulation of intragenic transposable elements impacts gene transcription in Arabidopsis thaliana. Nucleic Acids Res. 2015, 43, 3911–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.F.; Su, T.; Cheng, G.Q.; Wang, B.X.; Li, X.; Deng, C.L.; Gao, W.J. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes (Basel) 2017, 8, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Xin, Y.; Kuang, L.; He, N. Distribution and Characteristics of Transposable Elements in the Mulberry Genome. Plant Genome 2019, 12, 180094. [Google Scholar] [CrossRef] [Green Version]
- Roger, F.; Godhe, A.; Gamfeldt, L. Genetic Diversity and Ecosystem Functioning in the Face of Multiple Stressors. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Li, X.; Guo, K.; Zhu, X.; Chen, P.; Li, Y.; Xie, G.; Wang, L.; Wang, Y.; Persson, S.; Peng, L. Domestication of rice has reduced the occurrence of transposable elements within gene coding regions. BMC Genom. 2017, 18, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savolainen, O.; Pyhäjärvi, T.; Knürr, T. Gene Flow and Local Adaptation in Trees. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 595–619. [Google Scholar] [CrossRef]
- Eriksson, G. Pinus Sylvestris Recent Genetic Research; Swedish University of Agricultural Science: Uppsala, Sweden, 2008. [Google Scholar]
- Neale, D.B.; Ingvarsson, P.K. Population, quantitative and comparative genomics of adaptation in forest trees. Curr. Opin. Plant Biol. 2008, 11, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Prunier, J.; Verta, J.-P.; MacKay, J.J. Conifer genomics and adaptation: At the crossroads of genetic diversity and genome function. New Phytol. 2016, 209, 44–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, A.J.; van Heerwaarden, J.; Wegrzyn, J.L.; Nelson, C.D.; Ross-Ibarra, J.; González-Martínez, S.C.; Neale, D.B. Patterns of Population Structure and Environmental Associations to Aridity Across the Range of Loblolly Pine (Pinus taeda L., Pinaceae). Genetics 2010, 185, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Cumbie, W.P.; Eckert, A.; Wegrzyn, J.; Whetten, R.; Neale, D.; Goldfarb, B. Association genetics of carbon isotope discrimination, height and foliar nitrogen in a natural population of Pinus taeda L. Heredity (Edinb) 2011, 107, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Chhatre, V.E.; Byram, T.D.; Neale, D.B.; Wegrzyn, J.L.; Krutovsky, K.V. Genetic structure and association mapping of adaptive and selective traits in the east Texas loblolly pine (Pinus taeda L.) breeding populations. Tree Genet. Genomes 2013, 9, 1161–1178. [Google Scholar] [CrossRef]
- Lu, M.; Krutovsky, K.V.; Nelson, C.D.; Koralewski, T.E.; Byram, T.D.; Loopstra, C.A. Exome genotyping, linkage disequilibrium and population structure in loblolly pine (Pinus taeda L.). BMC Genom. 2016, 17, 730. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome/ Gene Set | Flanking Region from the Gene Start/End Coordinates | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | ||
| 0–1 kb | 0–1 kb | 1–2 kb | 1–2 kb | 2–3 kb | 2–3 kb | 3–4 kb | 3–4 kb | 4–5 kb | 4–5 kb | ||
| P. taeda v.2.0. all genes | Nb of extr.seq. | 36,726 | 36,728 | 34,711 | 34,063 | 33,184 | 32,310 | 31,767 | 30,838 | 30,349 | 29,479 |
| Nb of hqh to TE-dr | 5851 | 6450 | 4362 | 3901 | 3750 | 3628 | 3310 | 3069 | 3202 | 2924 | |
| ratio |  | ||||||||||
| >50 | 17 | 22 | 10 | 10 | 4 | 2 | 1 | 0 | 0 | 0 | |
| >100 | 8 | 9 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| P. taeda v.2.0. annotated genes | Nb of extr.seq. | 15,084 | 15,057 | 14,114 | 13,793 | 13,371 | 12,912 | 12,713 | 12,192 | 11,985 | 11,569 |
| Nb of hqh to TE-dr | 816 | 773 | 800 | 732 | 875 | 968 | 1161 | 991 | 901 | 1000 | |
| ratio |  | ||||||||||
| >50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| >100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| P. taeda v.1.01. HQ genes | Nb of extr.seq. | 4298 | 4239 | 4177 | 4128 | 4130 | 4091 | 4081 | 4028 | 4023 | 3967 |
| Nb of hqh to TE-dr | 784 | 779 | 2258 | 1890 | 3151 | 2693 | 3593 | 3222 | 3816 | 3539 | |
| ratio |  | ||||||||||
| >50 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| >100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| P. taeda v.1.01. LQ genes | Nb of extr.seq. | 75,425 | 75,459 | 72,840 | 72,797 | 71,554 | 71,470 | 70,002 | 69,836 | 68,237 | 68,017 |
| Nb of hqh to TE-dr | 2317 | 2540 | 4188 | 4243 | 4979 | 5070 | 5256 | 5387 | 5645 | 5382 | |
| ratio |  | ||||||||||
| >50 | 2 | 2 | 5 | 5 | 6 | 5 | 4 | 7 | 7 | 6 | |
| >100 | 1 | 1 | 3 | 4 | 1 | 1 | 0 | 1 | 0 | 0 | |
| P. lambertiana v.1.01 HQ genes | Nb of extr.seq. | 8779 | 8778 | 8746 | 8742 | 8719 | 8708 | 8692 | 8673 | 8660 | 8640 |
| Nb of hqh to TE-dr | 71 | 55 | 163 | 187 | 278 | 277 | 315 | 296 | 355 | 357 | |
| ratio |  | ||||||||||
| >50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| >100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| P. lambertiana v.1.01 LQ | Nb of extr.seq. | 71,162 | 71,157 | 70,386 | 70,475 | 69,773 | 69,909 | 69,217 | 69,344 | 68,660 | 68,836 |
| Nb of hqh to TE-dr | 470 | 466 | 1063 | 1011 | 1556 | 1508 | 1789 | 1368 | 2038 | 1999 | |
| ratio |  | ||||||||||
| >50 | 0 | 0 | 1 | 0 | 4 | 3 | 6 | 1 | 7 | 7 | |
| >100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Species | Genes ID with Multiple Plater MITEs | Nb of Plater Insertions | Description | qc, % | e-Value | ID, % | Accession |
|---|---|---|---|---|---|---|---|
| P. taeda v.2.0. | PITA_12742 | 7 | uncharacterized protein with domain of phosphoglucosamine mutase family protein | 88 | 0.00 × 100 | 65 | PLN02371 |
| PITA_21987 | 4 | subtilisin-like protease SBT5.3 | 96 | 0.0 | 47 | XP_012083905.1 | |
| PITA_00114 | 3 | metal tolerance protein 11 | 99 | 0.0 | 72 | XP_006857671.1 | |
| PITA_24114 | 2 | probable xyloglucan endotransglucosylase/hydrolase protein B | 93 | 3.00 × 10−153 | 72 | XP_030961064.1 | |
| PITA_21327 | 2 | 60S ribosomal protein L8-1-like | 95 | 2.00 × 10−169 | 90 | XP_022936671.1 | |
| PITA_17959 | 2 | TMV resistance protein N-like | 93 | 8.00 × 10−165 | 31 | XP_023886681.1 | |
| PITA_34859 | 2 | 3-oxoacyl-[acyl-carrier-protein] synthase I, chloroplastic-like isoform X1 | 95 | 0.0 | 74 | XP_028101593.1 | |
| PITA_28894 | 2 | L-gulonolactone oxidase 2 isoform X2 | 95 | 0.0 | 54 | XP_011621860.1 | |
| PITA_00539 | 2 | probable potassium transporter 11 | 99 | 0.0 | 67 | XP_006830082.1 | |
| PITA_33316 | 2 | plasma membrane intrinsic protein 2;8 | 95 | 5.00 × 10−141 | 74 | NP_179277.1 | |
| PITA_09881 | 2 | cytokinin hydroxylase | 93 | 0.0 | 52 | XP_011099558.1 | |
| P. lambertiana v.1.01. HQ genes | S/hiseq/c38458_g1_i1|m.23006 | 2 | bifunctional phosphatase IMPL2, chloroplastic | 75 | 7.00 × 10−147 | 73 | XP_011088446.1 |
| PILAhq_048992 | 2 | putative clathrin assembly protein At4g40080 | 80 | 2.00 × 10−40 | 36 | XP_027337607.1 | |
| PILAhm_002002 | 2 | histone deacetylase 15 isoform X3 | 69 | 4.00 × 10−179 | 63.89 | XP_010265267.1 |
| TE-dr Nb. Pita | TE-dr Nb. Pila | Description | Accession | Conserved Domain Name | Accession | GO Terms |
|---|---|---|---|---|---|---|
| 24 | 19 | plastidial pyruvate kinase 2 | XP_006843356.1 h | PLN02623 | PLN02623 | reproduction; ATP generation from ADP; seed maturation; |
| 23 | 26 | DEAD-box ATP-dependent RNA helicase 20 isoform X2/helicase 58, chloroplastic isoform X3 | XP_025888827.1 | SrmB | COG0513 | RNA secondary structure unwinding |
| 21 | 21 | phospholipid:diacylglycerol acyltransferase 1 | XP_006849611.1 h | PLN02517 | PLN02517 | acylglycerol biosynthetic process |
| 18 | 20 | nuclear pore complex protein NUP62-like/GPCR-type G protein 1 isoform X2 | XP_024396806.1 | SMC_prok_B super family | cl37069 | RNA export from nucleus; protein import/export into/from nucleus; nucleocytoplasmic transport, localization |
| 13 | 23 | WD repeat-containing protein WRAP73 | XP_008798782.1 | WD40 super family | COG2319 | - |
| - | 19 | protein RAE1 | XP_028076289.1 | cl29593 | ||
| - | 24 | actin-related protein 2/3 complex subunit 1A | XP_011627051.1 | cl29593 | ||
| 12 | 19 | uncharacterized protein LOC109715170/probable E3 ubiquitin-protein ligase HERC4 isoform X1 | XP_020095639.1 | ATS1 super family | cl34932 | - |
| 11 | 31 | peroxisomal adenine nucleotide carrier 1/mitochondrial substrate carrier family protein C-like | XP_006841423.1 | Mito_carr | pfam00153 | Establishment of localization; transmembrane transport; amide biosynthetic process; translation; nitrogen compound metabolic process. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voronova, A.; Rendón-Anaya, M.; Ingvarsson, P.; Kalendar, R.; Ruņģis, D. Comparative Study of Pine Reference Genomes Reveals Transposable Element Interconnected Gene Networks. Genes 2020, 11, 1216. https://doi.org/10.3390/genes11101216
Voronova A, Rendón-Anaya M, Ingvarsson P, Kalendar R, Ruņģis D. Comparative Study of Pine Reference Genomes Reveals Transposable Element Interconnected Gene Networks. Genes. 2020; 11(10):1216. https://doi.org/10.3390/genes11101216
Chicago/Turabian StyleVoronova, Angelika, Martha Rendón-Anaya, Pär Ingvarsson, Ruslan Kalendar, and Dainis Ruņģis. 2020. "Comparative Study of Pine Reference Genomes Reveals Transposable Element Interconnected Gene Networks" Genes 11, no. 10: 1216. https://doi.org/10.3390/genes11101216
APA StyleVoronova, A., Rendón-Anaya, M., Ingvarsson, P., Kalendar, R., & Ruņģis, D. (2020). Comparative Study of Pine Reference Genomes Reveals Transposable Element Interconnected Gene Networks. Genes, 11(10), 1216. https://doi.org/10.3390/genes11101216