DNA Damage and Repair in Pulmonary Arterial Hypertension



Abstract
:1. Introduction
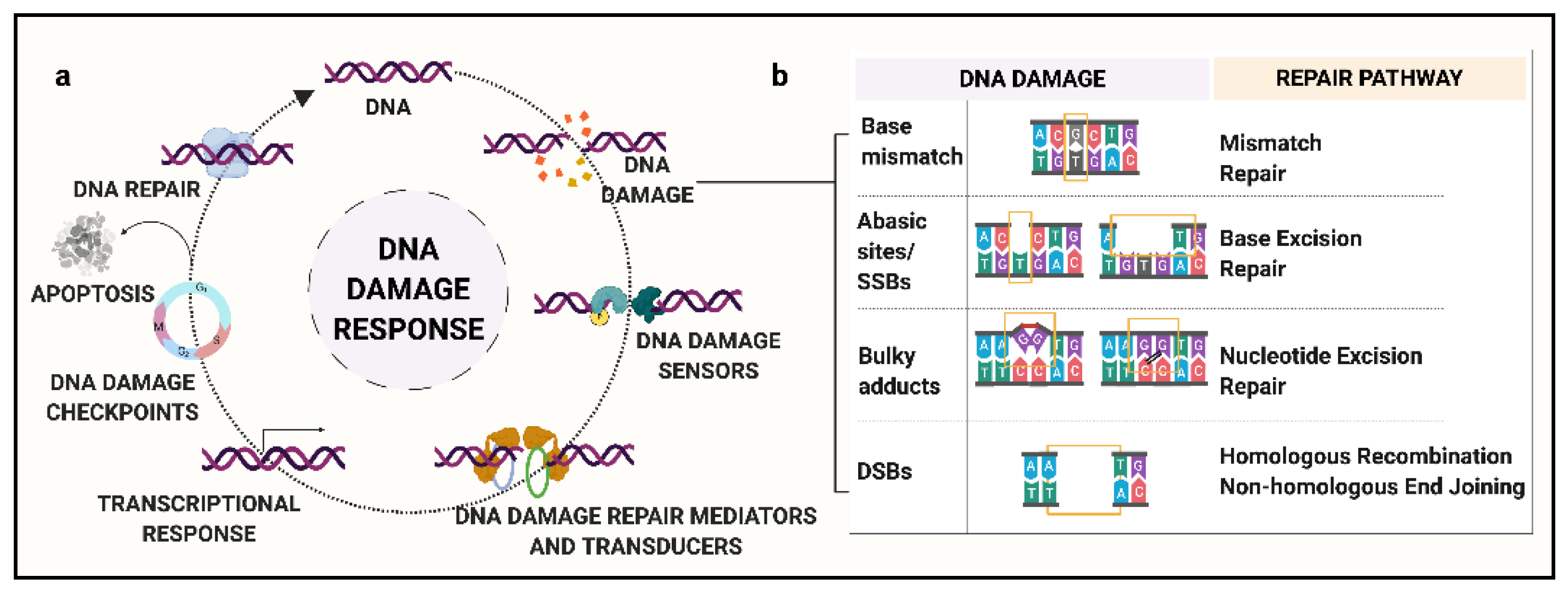
2. Background: DNA Damage and Response Pathways
- (a)
- Mismatch Repair (MMR): MMR is responsible for the recognition and repair of base mismatches. Base mismatches can arise as a result of covalent or non-covalent structural changes, or due to insertion/deletions resulting from replication errors or recombination [28]. For example, methylated guanine base, O6MeGua, has a high frequency of pairing to thymine (T), activating MMR to excise the mismatched T residue. Loss of MMR can lead to a significant increase in spontaneous mutations. Major known genes in the MMR pathway include MGMT, MSH6, and MLH3.
- (b)
- Base Excision Repair (BER): This repair process is governed by DNA glycosylases along with endonucleases that recognize and eliminate the modified or damaged bases, such as oxidized, reduced, alkylated or deaminated bases, to generate an abasic site [29]. For example, in humans, 8-oxoguanine glycosylase-1 (OGG1) recognizes and removes the oxidatively modified guanine base, 8-oxoGuanine (8-oxoG) via incision of the 3′-phosphodiester bond. Following this step, the apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1) cleaves the 5′-bond generating a 1-nt abasic site [30]. Major genes of the BER pathway include MBD4, OGG1, MUTYH, and NEIL1.
- (c)
- Nucleotide Excision Repair (NER): Unlike BER, NER involves a complex of enzymes that work in coordination to recognize SSBs and remove bulky lesions [31]. Briefly, the steps include recognition of the damaged site, a dual incision at extreme ends of the lesion, elimination of damaged oligomer, and new base synthesis followed by ligation [32]. Major known NER genes include XPC, XPA, and ERCC1-5.
- (d)
- Homologous Recombination (HR): As compared to the excision repair pathways, HR is a far more complex phenomenon. HR involves multiple-step processing of DSBs by several different proteins with specific functions [33]. The key characteristic of HR is that it uses a homologous duplex template to retrieve the lost information. It is a complex phenomenon, with the potential for incorrect template usage that can lead to gene conversion. Major genes involved in the HR pathway include RAD51, BRCA1, BRCA2, and the Mre11/Rad50/NBS1 complex [34].
- (e)
- Non-Homologous End Joining (NHEJ): Similar to HR, NHEJ involves multiple-step repair processing of DSBs. In this mechanism, the two ends of DSBs are stabilized by DNA-protein kinases and ligated together [35]. It is believed to be the main repair pathway for DSBs induced by ionizing radiation. Major proteins implicated in NHEJ include KU70/80 heterodimer and XRCC4 [36,37]. A lack of specific recognition criteria for the ligated ends can lead to erroneous joining of non-contiguous DNA sequences, giving rise to structural rearrangements.
3. DNA Damage and Genetic Instability in PAH
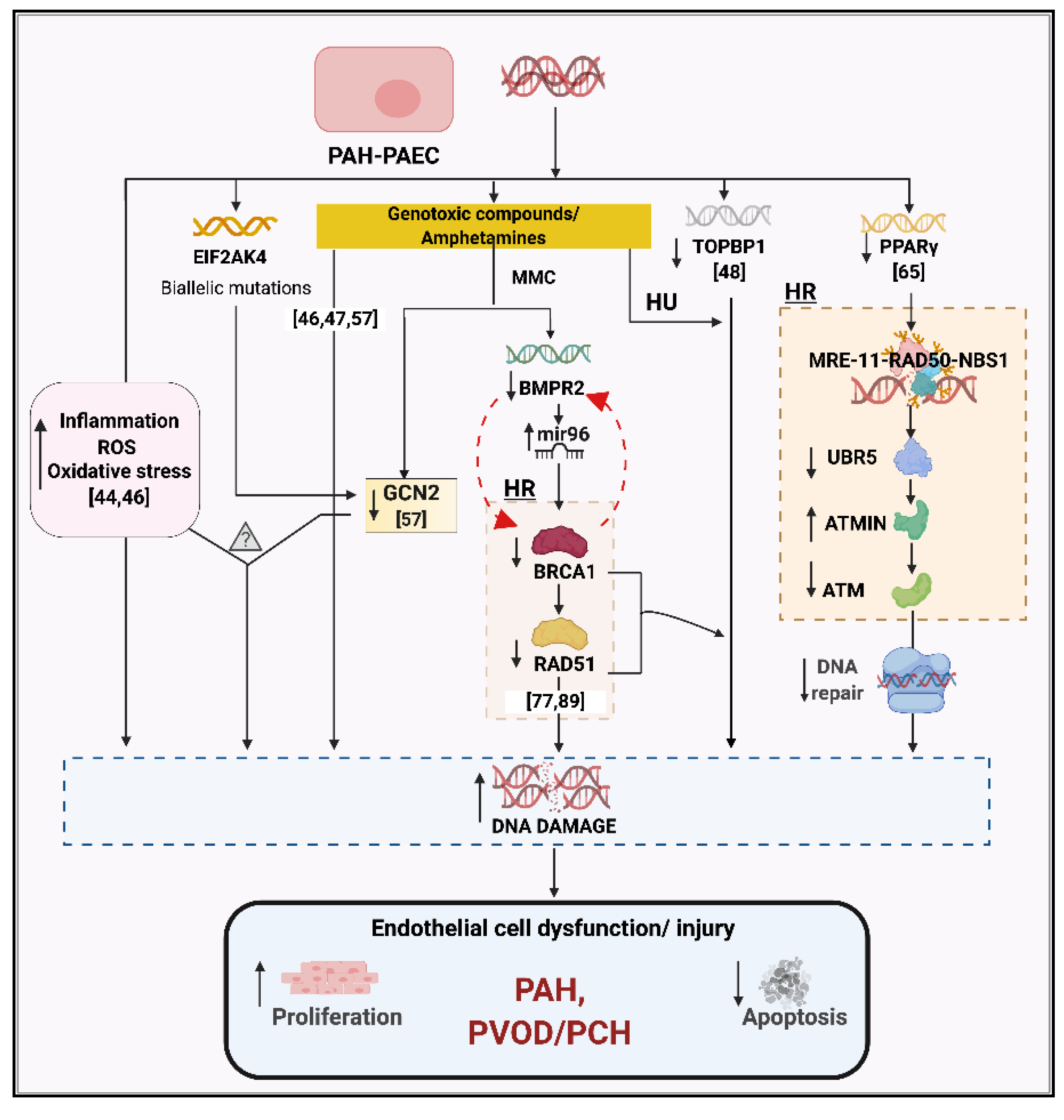
4. Role of Mutagens and Environmental Modifiers
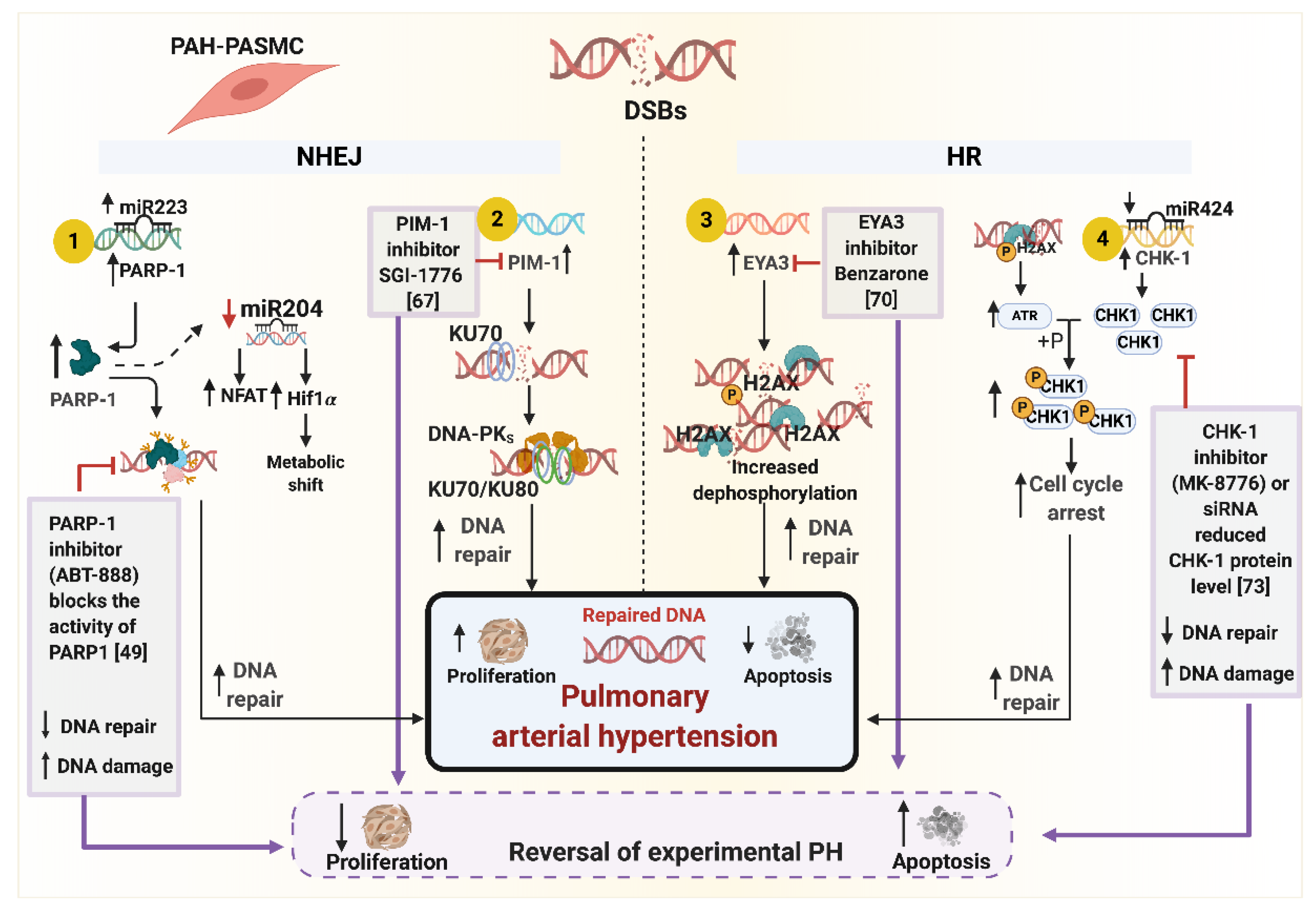
5. DNA Repair Pathways and Cell Cycle Checkpoints in PAH
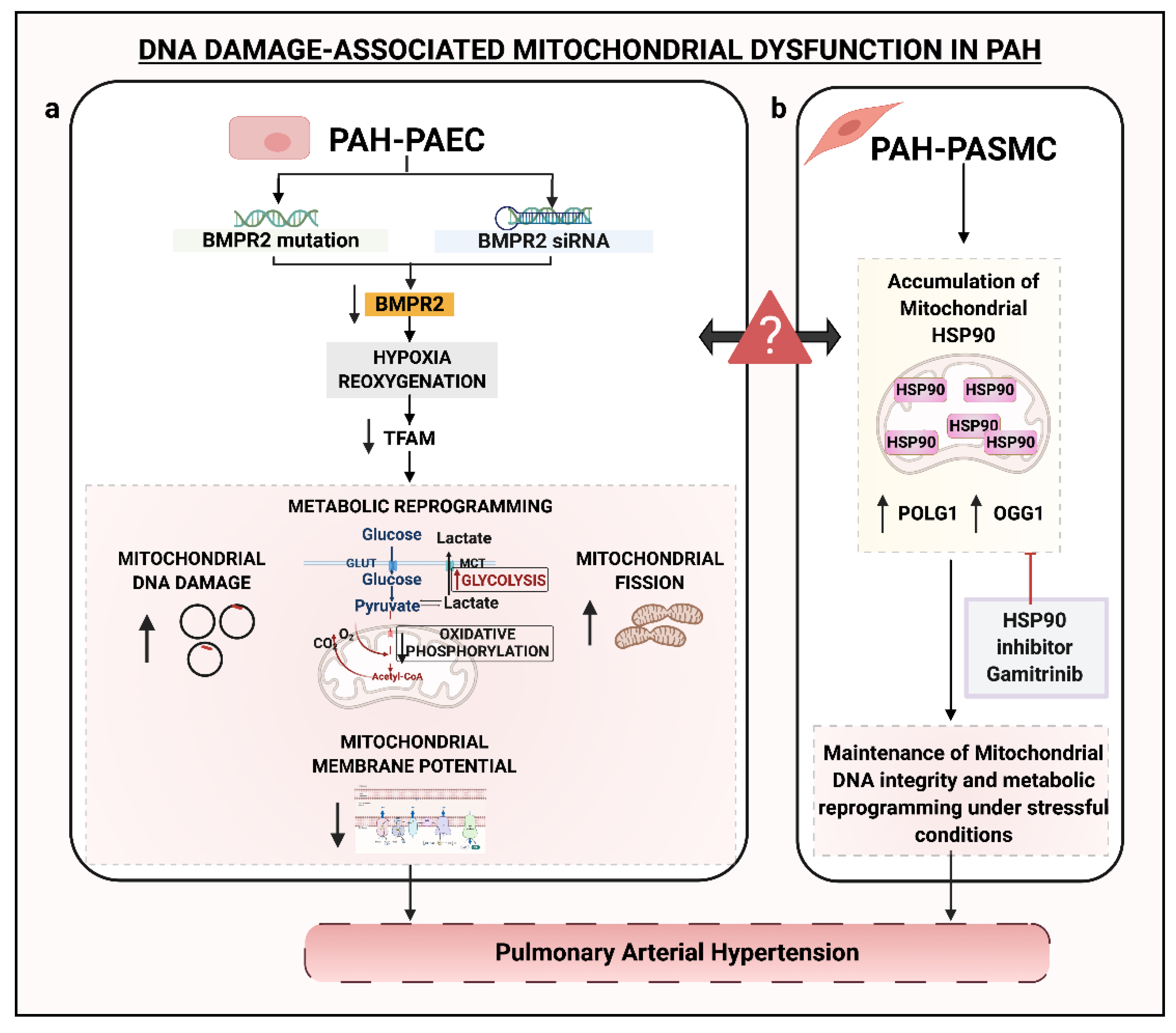
6. DNA Damage and Mitochondria
7. BMPR2 and DNA Damage
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.C.; Gibbs, J.S. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Montani, D.; Perros, F.; Dorfmuller, P.; Adnot, S.; Eddahibi, S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul. Pharmacol. 2008, 49, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [Green Version]
- Potus, F.; Graydon, C.; Provencher, S.; Bonnet, S. Vascular remodeling process in pulmonary arterial hypertension, with focus on miR-204 and miR-126 (2013 Grover Conference series). Pulm. Circ. 2014, 4, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Hoeper, M.M.; McLaughlin, V.V.; Dalaan, A.M.; Satoh, T.; Galie, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [Green Version]
- International, P.P.H.C.; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, M.; Yagi, H.; Nakayama, T.; Saji, T.; Matsuoka, R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 2009, 46, 331–337. [Google Scholar] [CrossRef]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J. Am. Coll. Cardiol. 2009, 53, 1573–1619. [Google Scholar] [CrossRef]
- Loyd, J.E. Pulmonary arterial hypertension: Insights from genetic studies. Proc. Am. Thorac. Soc. 2011, 8, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Demarco, V.G.; Whaley-Connell, A.T.; Sowers, J.R.; Habibi, J.; Dellsperger, K.C. Contribution of oxidative stress to pulmonary arterial hypertension. World J. Cardiol. 2010, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Kherbeck, N.; Tamby, M.C.; Bussone, G.; Dib, H.; Perros, F.; Humbert, M.; Mouthon, L. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin. Rev. Allergy Immunol. 2013, 44, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaminsky, S.; Hautefort, A.; Price, L.; Humbert, M.; Perros, F. Inflammation in pulmonary hypertension: What we know and what we could logically and safely target first. Drug Discov. Today 2014, 19, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B. Pulmonary Arterial Hypertension Is Associated with Oxidative Stress-induced Genome Instability. Am. J. Respir. Crit. Care Med. 2015, 192, 129–130. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Izumi, T.; Wiederhold, L.R.; Roy, G.; Roy, R.; Jaiswal, A.; Bhakat, K.K.; Mitra, S.; Hazra, T.K. Mammalian DNA base excision repair proteins: Their interactions and role in repair of oxidative DNA damage. Toxicology 2003, 193, 43–65. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Sancar, A. DNA excision repair. Annu. Rev. Biochem. 1996, 65, 43–81. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.D. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 1999, 81, 39–44. [Google Scholar] [CrossRef]
- Modesti, M.; Kanaar, R. Homologous recombination: From model organisms to human disease. Genome Biol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R., 3rd; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, T.M.; Jackson, S.P. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef]
- Ramsden, D.A.; Gellert, M. Ku protein stimulates DNA end joining by mammalian DNA ligases: A direct role for Ku in repair of DNA double-strand breaks. EMBO J. 1998, 17, 609–614. [Google Scholar] [CrossRef] [Green Version]
- Nick McElhinny, S.A.; Snowden, C.M.; McCarville, J.; Ramsden, D.A. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell. Biol. 2000, 20, 2996–3003. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006, 40, 209–235. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Radisavljevic, Z.; Shroyer, K.R.; Polak, J.M.; Voelkel, N.F. Monoclonal endothelial cells in appetite suppressant-associated pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 1999–2001. [Google Scholar] [CrossRef]
- Lee, S.D.; Shroyer, K.R.; Markham, N.E.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J. Clin. Investig. 1998, 101, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Voelkel, N.F.; Cool, C.; Lee, S.D.; Wright, L.; Geraci, M.W.; Tuder, R.M. Primary pulmonary hypertension between inflammation and cancer. Chest 1998, 114, 225S–230S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, M.E.; Halley, G.R.; Golpon, H.A.; Voelkel, N.F.; Tuder, R.M. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ. Res. 2001, 88, E2–E11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.D.; James, V.; Southwood, M.; Harrison, R.E.; Atkinson, C.; Stewart, S.; Morrell, N.W.; Trembath, R.C.; Aldred, M.A. Investigation of second genetic hits at the BMPR2 locus as a modulator of disease progression in familial pulmonary arterial hypertension. Circulation 2005, 111, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.M.; Comhair, S.A.; Erzurum, S.C.; Tuder, R.M.; Aldred, M.A. Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dysregulates SMAD9 signaling. Am. J. Respir. Crit. Care Med. 2015, 191, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Federici, C.; Drake, K.M.; Rigelsky, C.M.; McNelly, L.N.; Meade, S.L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.I.; Cao, A.; Miyagawa, K.; Tojais, N.F.; Hennigs, J.K.; Li, C.G.; Sweeney, N.M.; Inglis, A.S.; Wang, L.; Li, D.; et al. Amphetamines promote mitochondrial dysfunction and DNA damage in pulmonary hypertension. JCI Insight 2017, 2, e90427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jesus Perez, V.A.; Yuan, K.; Lyuksyutova, M.A.; Dewey, F.; Orcholski, M.E.; Shuffle, E.M.; Mathur, M.; Yancy, L., Jr.; Rojas, V.; Li, C.G.; et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.; Ali, M.; McCallum, G.P.; Kumareswaran, R.; Koritzinsky, M.; Wouters, B.G.; Wells, P.G.; Gallinger, S.; Bristow, R.G. Hypoxia provokes base excision repair changes and a repair-deficient, mutator phenotype in colorectal cancer cells. Mol. Cancer Res. 2014, 12, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef] [Green Version]
- Hammond, E.M.; Dorie, M.J.; Giaccia, A.J. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J. Biol. Chem. 2003, 278, 12207–12213. [Google Scholar] [CrossRef] [Green Version]
- Scanlon, S.E.; Glazer, P.M. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst.) 2015, 32, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jesus Perez, V.; Kudelko, K.; Snook, S.; Zamanian, R.T. Drugs and toxins-associated pulmonary arterial hypertension: Lessons learned and challenges ahead. Int. J. Clin. Pract. Suppl. 2011, 8–10. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Raudensky, J. The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J. Neuroimmune Pharmacol. 2008, 3, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, C.J.; Batai, K.; Bleda, M.; Haimel, M.; Southgate, L.; Germain, M.; Pauciulo, M.W.; Hadinnapola, C.; Aman, J.; Girerd, B.; et al. Genetic determinants of risk in pulmonary arterial hypertension: International genome-wide association studies and meta-analysis. Lancet Respir. Med. 2019, 7, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Perros, F.; Gunther, S.; Ranchoux, B.; Godinas, L.; Antigny, F.; Chaumais, M.C.; Dorfmuller, P.; Hautefort, A.; Raymond, N.; Savale, L.; et al. Mitomycin-Induced Pulmonary Veno-Occlusive Disease: Evidence From Human Disease and Animal Models. Circulation 2015, 132, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.J.; Bunpo, P.; Cundiff, J.K.; Wek, R.C.; Anthony, T.G. The eukaryotic initiation factor 2 kinase GCN2 protects against hepatotoxicity during asparaginase treatment. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1124–E1133. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmuller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65–69. [Google Scholar] [CrossRef]
- Best, D.H.; Sumner, K.L.; Austin, E.D.; Chung, W.K.; Brown, L.M.; Borczuk, A.C.; Rosenzweig, E.B.; Bayrak-Toydemir, P.; Mao, R.; Cahill, B.C.; et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014, 145, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Drake, K.M.; Federici, C.; Duong, H.T.; Comhair, S.A.; Erzurum, S.C.; Asosingh, K.; Aldred, M.A. Genomic stability of pulmonary artery endothelial colony-forming cells in culture. Pulm. Circ. 2017, 7, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Guignabert, C.; Alvira, C.M.; Alastalo, T.P.; Sawada, H.; Hansmann, G.; Zhao, M.; Wang, L.; El-Bizri, N.; Rabinovitch, M. Tie2-mediated loss of peroxisome proliferator-activated receptor-γ in mice causes PDGF receptor-β-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1082–L1090. [Google Scholar] [CrossRef] [Green Version]
- Alastalo, T.P.; Li, M.; Perez Vde, J.; Pham, D.; Sawada, H.; Wang, J.K.; Koskenvuo, M.; Wang, L.; Freeman, B.A.; Chang, H.Y.; et al. Disruption of PPARgamma/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J. Clin. Investig. 2011, 121, 3735–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.G.; Mahon, C.; Sweeney, N.M.; Verschueren, E.; Kantamani, V.; Li, D.; Hennigs, J.K.; Marciano, D.P.; Diebold, I.; Abu-Halawa, O.; et al. PPARgamma Interaction with UBR5/ATMIN Promotes DNA Repair to Maintain Endothelial Homeostasis. Cell Rep. 2019, 26, 1333–1343.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloche, J.; Le Guen, M.; Potus, F.; Vinck, J.; Ranchoux, B.; Johnson, I.; Antigny, F.; Tremblay, E.; Breuils-Bonnet, S.; Perros, F.; et al. miR-223 reverses experimental pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2015, 309, C363–C372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampron, M.C.; Vitry, G.; Nadeau, V.; Grobs, Y.; Paradis, R.; Samson, N.; Tremblay, E.; Boucherat, O.; Meloche, J.; Bonnet, S.; et al. PIM1 (Moloney Murine Leukemia Provirus Integration Site) Inhibition Decreases the Nonhomologous End-Joining DNA Damage Repair Signaling Pathway in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 783–801. [Google Scholar] [CrossRef]
- Hsu, J.L.; Leong, P.K.; Ho, Y.F.; Hsu, L.C.; Lu, P.H.; Chen, C.S.; Guh, J.H. Pim-1 knockdown potentiates paclitaxel-induced apoptosis in human hormone-refractory prostate cancers through inhibition of NHEJ DNA repair. Cancer Lett. 2012, 319, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef]
- Wang, Y.; Pandey, R.N.; York, A.J.; Mallela, J.; Nichols, W.C.; Hu, Y.C.; Molkentin, J.D.; Wikenheiser-Brokamp, K.A.; Hegde, R.S. The EYA3 tyrosine phosphatase activity promotes pulmonary vascular remodeling in pulmonary arterial hypertension. Nat. Commun. 2019, 10, 4143. [Google Scholar] [CrossRef] [Green Version]
- Carrassa, L.; Broggini, M.; Erba, E.; Damia, G. Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: Differential activity in cells expressing or not p53. Cell Cycle 2004, 3, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, A.; Bonnet, S.; Breuils-Bonnet, S.; Habbout, K.; Paradis, R.; Tremblay, E.; Lampron, M.C.; Orcholski, M.E.; Potus, F.; Bertero, T.; et al. Inhibition of CHK 1 (Checkpoint Kinase 1) Elicits Therapeutic Effects in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Vattulainen-Collanus, S.; Southwood, M.; Yang, X.D.; Moore, S.; Ghatpande, P.; Morrell, N.W.; Lagna, G.; Hata, A. Bone morphogenetic protein signaling is required for RAD51-mediated maintenance of genome integrity in vascular endothelial cells. Commun. Biol. 2018, 1, 149. [Google Scholar] [CrossRef] [Green Version]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Courboulin, A.; Paulin, R.; Giguere, N.J.; Saksouk, N.; Perreault, T.; Meloche, J.; Paquet, E.R.; Biardel, S.; Provencher, S.; Cote, J.; et al. Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 2011, 208, 535–548. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef]
- Fessel, J.P.; West, J.D. Redox biology in pulmonary arterial hypertension (2013 Grover Conference Series). Pulm. Circ. 2015, 5, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikal, A.A. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark. Med. 2010, 4, 241–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 Accumulation Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103. [Google Scholar] [CrossRef]
- Li, M.; Vattulainen, S.; Aho, J.; Orcholski, M.; Rojas, V.; Yuan, K.; Helenius, M.; Taimen, P.; Myllykangas, S.; De Jesus Perez, V.; et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1118–1128. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.W.; Calses, P.; Kemp, C.J.; Taniguchi, T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012, 72, 4037–4046. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Damage and Response Genes | PAH-PAECs | PAH-PASMCs |
|---|---|---|
| Base Excision Repair | ||
| OGG1 (8-Oxoguanine DNA Glycosylase) | Not known | Increased expression [88] |
| Reduced expression ** [67] | ||
| Homologous recombination | ||
| RAD51 (RAD51 Recombinase) | Reduced expression [77] | Increased expression [73] |
| BRCA1 (Breast and Ovarian Cancer Susceptibility Protein 1) | Reduced expression # [77] | Reduced expression [89] |
| NBS1 (Nibrin) | Not known | Reduced expression *** |
| Non-homologous end-joining | ||
| XRCC6 (Ku70) (X-Ray Repair Cross Complementing 6) | Not known | Reduced expression ** |
| PARP-1 (Poly (ADP-Ribose) Polymerase 1) | Not known | Increased expression [49] |
| Other genes involved in regulation of DNA damage | ||
| BMPR2 | Reduced expression # [89] | No change in expression * [89] |
| TFAM | Reduced expression [78] | Not known |
| TOPBP1 (DNA Topoisomerase II Binding Protein 1) | Reduced expression [48] | Not known |
| PPARG-UBR5 | No change in expression but reduced interaction observed [65] | Not known |
| ATMIN | Increased expression [65] | Not known |
| PIM1 | Not known | Increased expression [67] |
| EYA3 | Not known | Increased expression [70] |
| CHK1 (Check point Kinase-1) | No association [73] | Increased expression [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Aldred, M.A. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes 2020, 11, 1224. https://doi.org/10.3390/genes11101224
Sharma S, Aldred MA. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes. 2020; 11(10):1224. https://doi.org/10.3390/genes11101224
Chicago/Turabian StyleSharma, Samantha, and Micheala A. Aldred. 2020. "DNA Damage and Repair in Pulmonary Arterial Hypertension" Genes 11, no. 10: 1224. https://doi.org/10.3390/genes11101224
APA StyleSharma, S., & Aldred, M. A. (2020). DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes, 11(10), 1224. https://doi.org/10.3390/genes11101224