Structural and Biochemical Features of Eimeria tenella Dihydroorotate Dehydrogenase, a Potential Drug Target

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Analysis of E. tenella DHODH
2.2. Expression and Purification of EtDHODH, PfDHODH, and HsDHODH
2.3. Enzyme Assays and Protein Concentration Determination
2.4. Screening of Inhibitors
2.5. Crystallization and X-ray Diffraction Data Collection for EtDHODH
2.6. Structural Determination of EtDHODH with and without Inhibitor
2.7. Structure Determination of HsDHODH-Ferulenol Complex
3. Results
3.1. Sequence Analysis of EtDHODH
3.2. Purification of DHODHs
3.3. Screening for EtDHODH Inhibitors
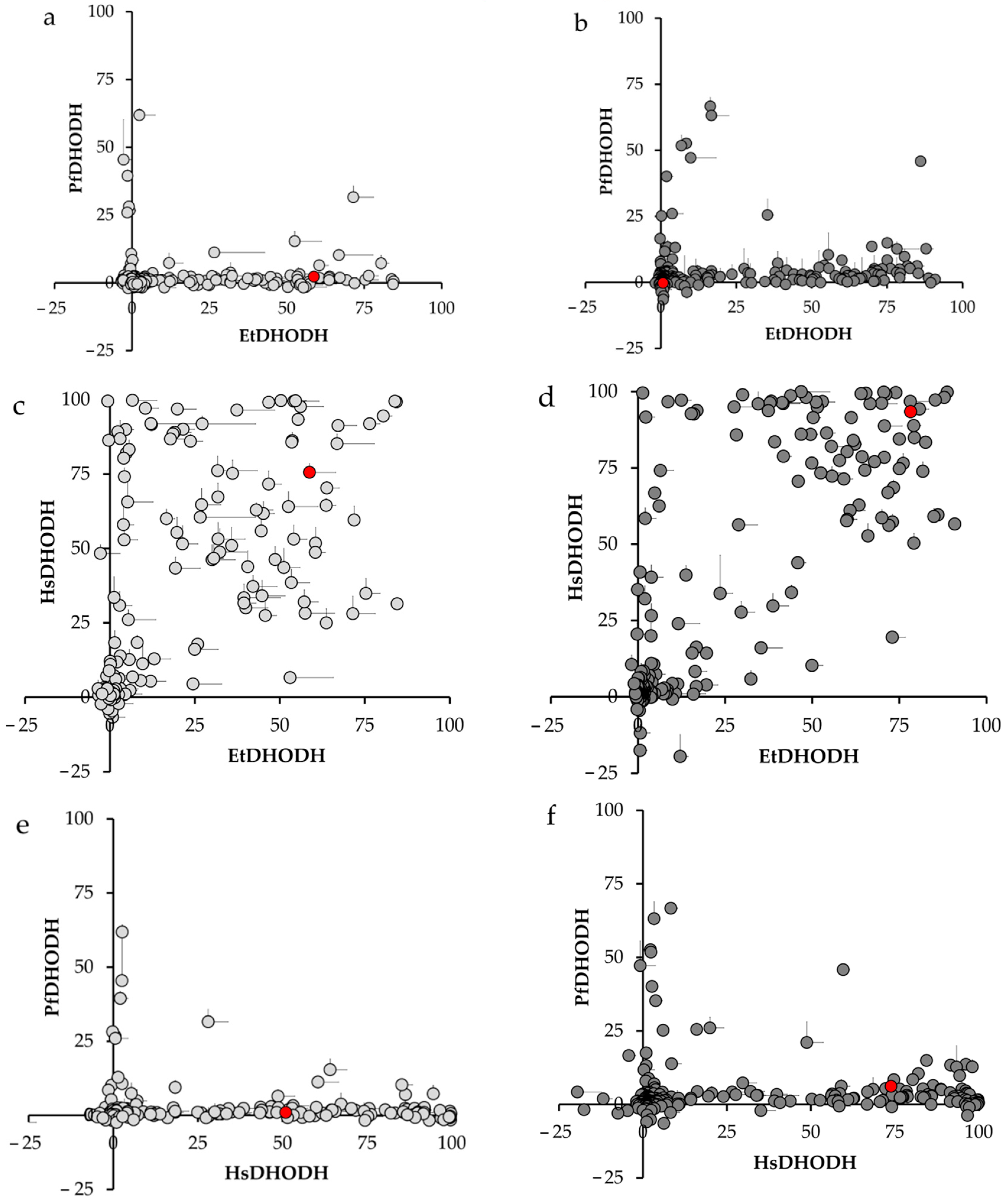
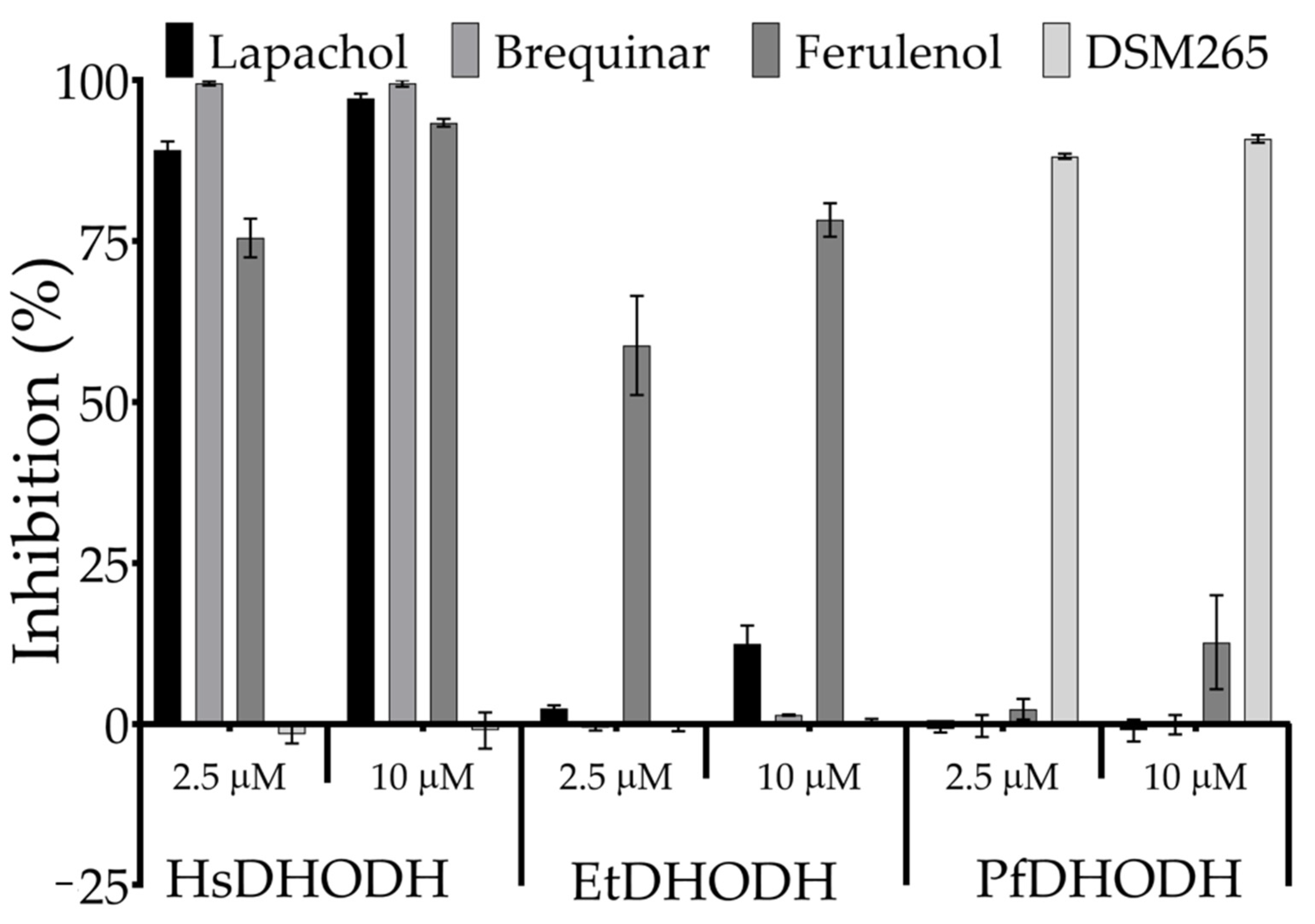
3.4. Inhibitor Cross-Sensitivity of EtDHODH, PfDHODH, and HsDHODH
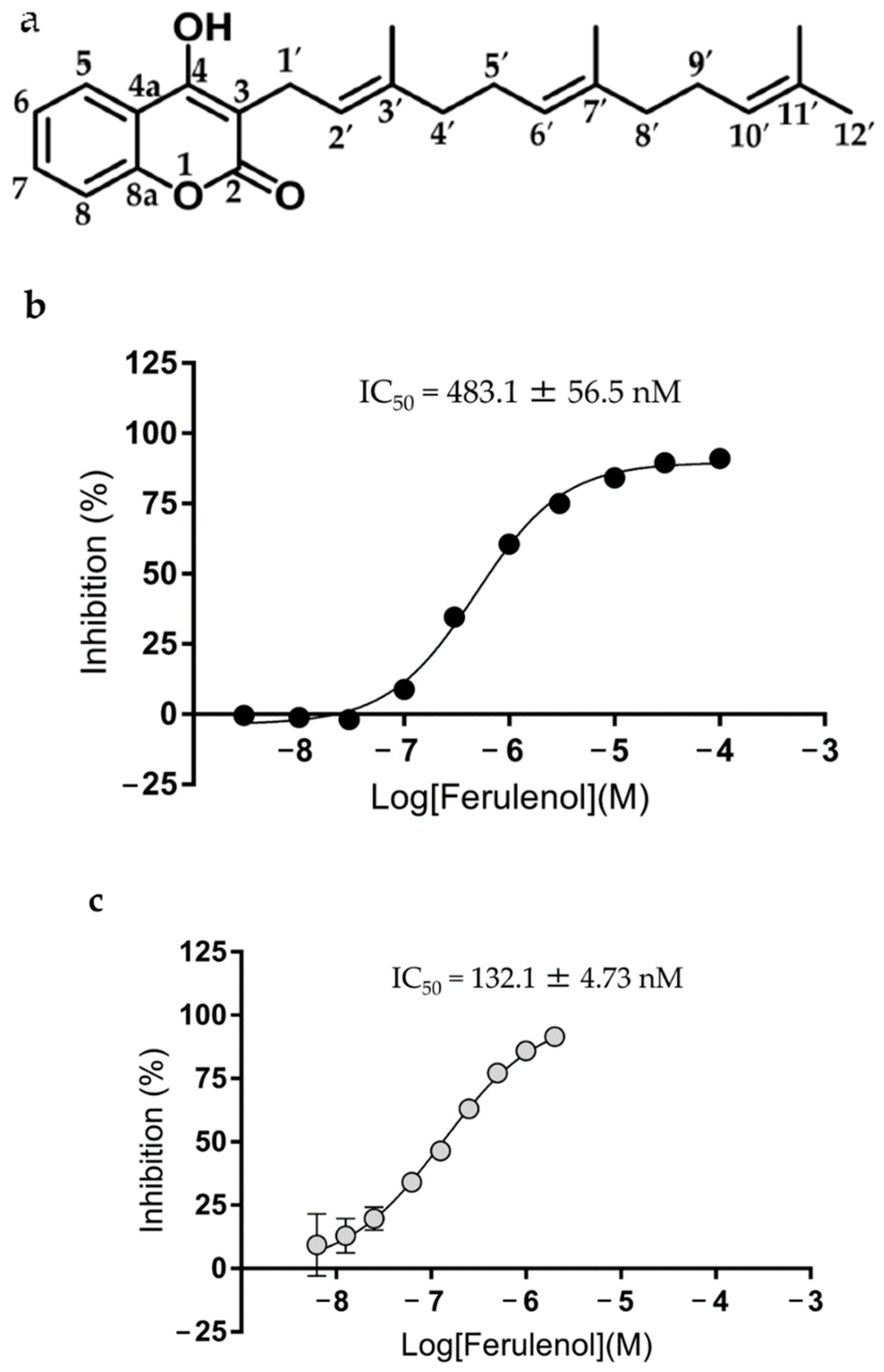
3.5. Ferulenol is a Potent Inhibitor of EtDHODH and HsDHODH
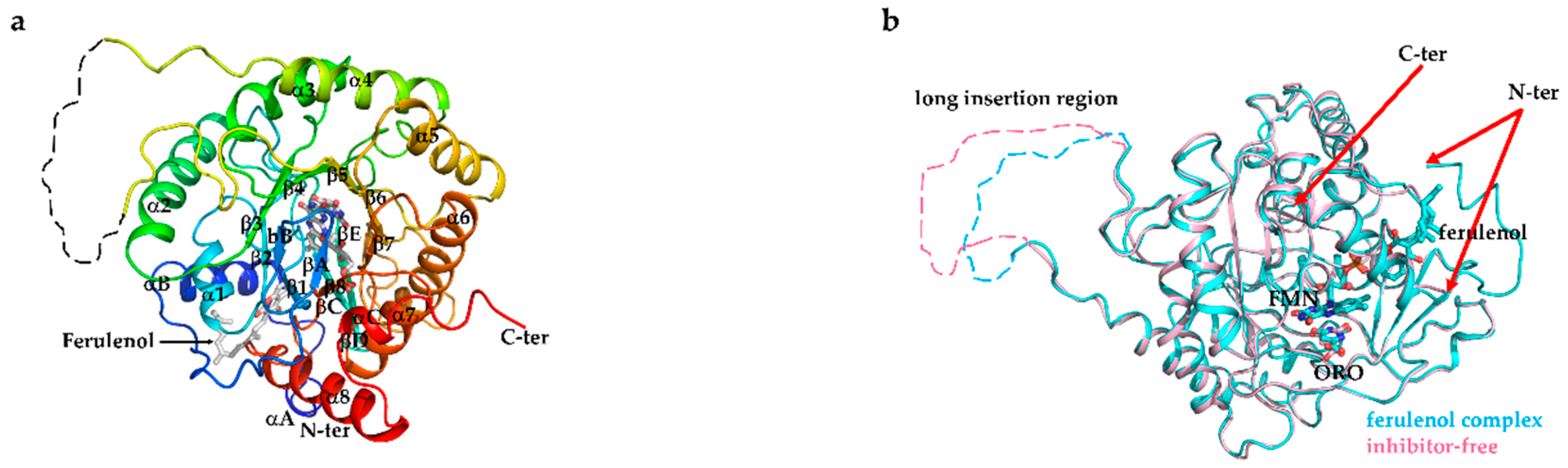
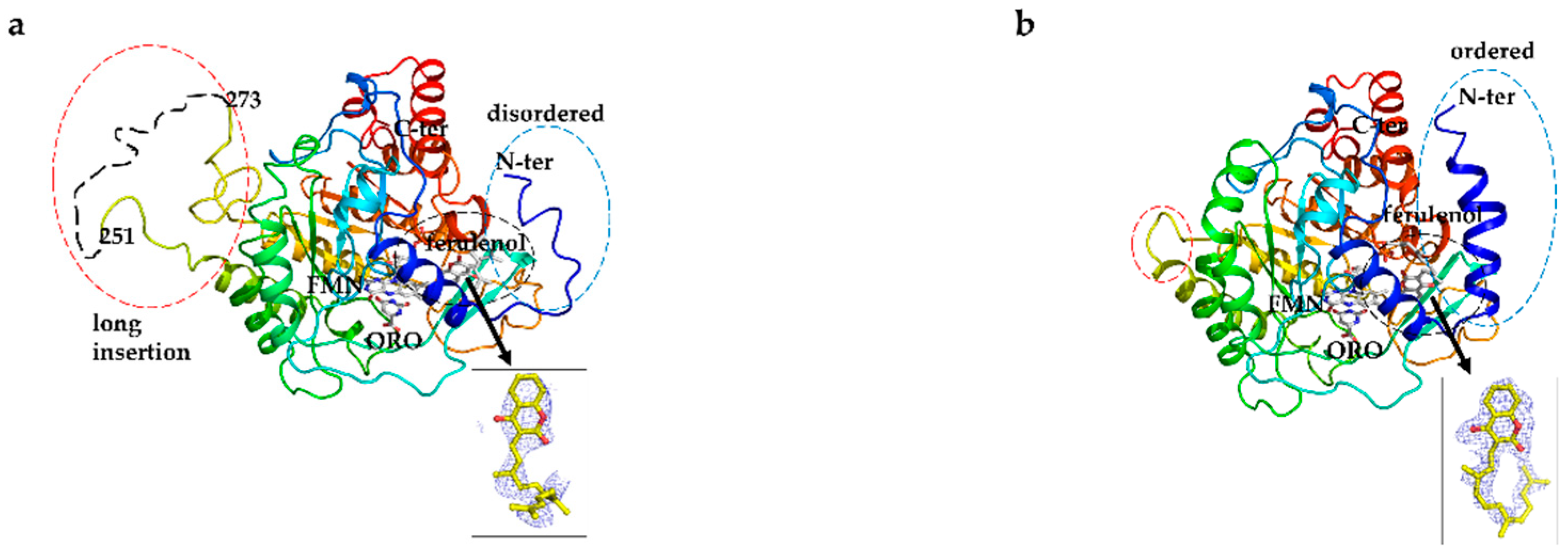
3.6. Overall Structure of EtDHODH and Comparison to HsDHODH Structure
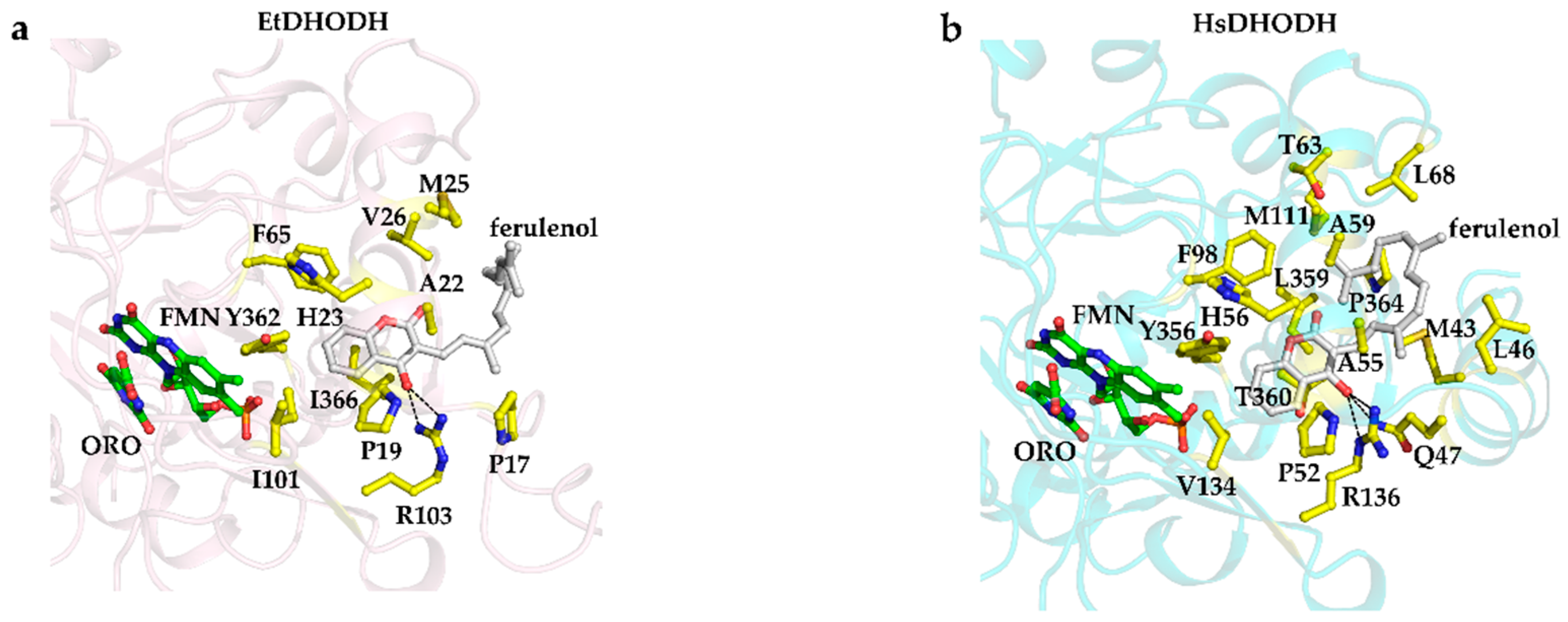
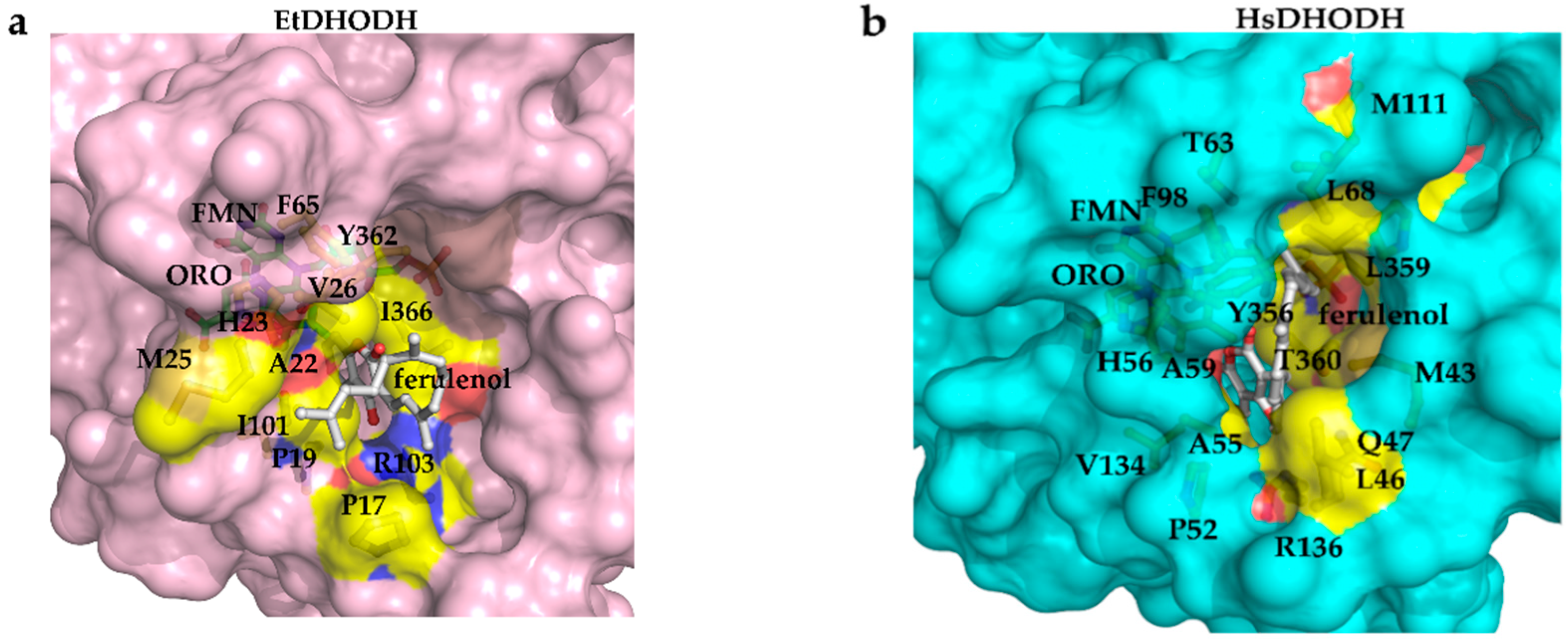
3.7. Recognition of Ferulenol by EtDHODH and HsDHODH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allen, P.C.; Fetterer, R.H. Recent Advances in Biology and Immunobiology of Eimeria Species and in Diagnosis and Control of Infection with These Coccidian Parasites of Poultry. Clin. Microbiol. Rev. 2002, 15, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, H.D.; Barta, J.R.; Blake, D.; Gruber, A.; Jenkins, M.; Smith, N.C.; Suo, X.; Tomley, F.M. A selective review of advances in coccidiosis research. Adv. Parasitol. 2013, 83, 93–171. [Google Scholar] [CrossRef] [PubMed]
- Witcombe, D.M.; Smith, N.C. Strategies for anti-coccidial prophylaxis. Parasitology 2014, 141, 1379–1389. [Google Scholar] [CrossRef]
- Birth, D.; Kao, W.C.; Hunte, C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.D.; Siregar, J.E.; Mollard, V.; Vega-Rodriguez, J.; Syafruddin, D.; Matsuoka, H.; Matsuzaki, M.; Toyama, T.; Sturm, A.; Cozijnsen, A.; et al. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 2016, 352, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Siregar, J.E.; Kurisu, G.; Kobayashi, T.; Matsuzaki, M.; Sakamoto, K.; Mi-ichi, F.; Watanabe, Y.; Hirai, M.; Matsuoka, H.; Syafruddin, D.; et al. Direct evidence for the atovaquone action on the Plasmodium cytochrome bc1 complex. Parasitol. Int. 2015, 64, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Ke, H.; Lewis, I.A.; Morrisey, J.M.; McLean, K.J.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Jacobs-Lorena, M.; Llinas, M.; Vaidya, A.B. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 2015, 11, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Mogi, T.; Kita, K. Diversity in mitochondrial metabolic pathways in parasitic protists Plasmodium and Cryptosporidium. Parasitol. Int. 2010, 59, 305–312. [Google Scholar] [CrossRef]
- Boysen, K.E.; Matuschewski, K. Arrested oocyst maturation in Plasmodium parasites lacking type II NADH:ubiquinone dehydrogenase. J. Biol. Chem. 2011, 286, 32661–32671. [Google Scholar] [CrossRef] [Green Version]
- Ke, H.; Ganesan, S.M.; Dass, S.; Morrisey, J.M.; Pou, S.; Nilsen, A.; Riscoe, M.K.; Mather, M.W.; Vaidya, A.B. Mitochondrial type II NADH dehydrogenase of Plasmodium falciparum (PfNDH2) is dispensable in the asexual blood stages. PLoS ONE 2019, 14, e0214023. [Google Scholar] [CrossRef] [Green Version]
- Hino, A.; Hirai, M.; Tanaka, T.Q.; Watanabe, Y.; Matsuoka, H.; Kita, K. Critical roles of the mitochondrial complex II in oocyst formation of rodent malaria parasite Plasmodium berghei. J. Biochem 2012, 152, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.Q.; Hirai, M.; Watanabe, Y.; Kita, K. Toward understanding the role of mitochondrial complex II in the intraerythrocytic stages of Plasmodium falciparum: Gene targeting of the Fp subunit. Parasitol. Int. 2012, 61, 726–728. [Google Scholar] [CrossRef]
- Niikura, M.; Komatsuya, K.; Inoue, S.I.; Matsuda, R.; Asahi, H.; Inaoka, D.K.; Kita, K.; Kobayashi, F. Suppression of experimental cerebral malaria by disruption of malate:quinone oxidoreductase. Malar. J. 2017, 16, 247. [Google Scholar] [CrossRef] [Green Version]
- Lian, L.Y.; Al-Helal, M.; Roslaini, A.M.; Fisher, N.; Bray, P.G.; Ward, S.A.; Biagini, G.A. Glycerol: An unexpected major metabolite of energy metabolism by the human malaria parasite. Malar. J. 2009, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Olszewski, K.L.; Llinas, M. Central carbon metabolism of Plasmodium parasites. Mol. Biochem. Parasitol. 2011, 175, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaoka, D.K.; Sakamoto, K.; Shimizu, H.; Shiba, T.; Kurisu, G.; Nara, T.; Aoki, T.; Kita, K.; Harada, S. Structures of Trypanosoma cruzi dihydroorotate dehydrogenase complexed with substrates and products: Atomic resolution insights into mechanisms of dihydroorotate oxidation and fumarate reduction. Biochemistry 2008, 47, 10881–10891. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Inaoka, D.K.; Shiba, T.; Saimoto, H.; Sakura, T.; Amalia, E.; Kido, Y.; Sakai, C.; Nakamura, M.; Moore, A.L.; et al. Selective Cytotoxicity of Dihydroorotate Dehydrogenase Inhibitors to Human Cancer Cells Under Hypoxia and Nutrient-Deprived Conditions. Front. Pharm. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.; Kilsby, J.; Rogerson, G.W.; McIntosh, R.T.; Ginger, C.D. The enzymes of pyrimidine biosynthesis in a range of parasitic protozoa and helminths. Mol. Biochem. Parasitol. 1981, 2, 123–134. [Google Scholar] [CrossRef]
- Khovanskikh, A.E. The assimilation by Eimeria tenella coccidia of DNA and RNA precursors from the host cell. Parazitologiia 1979, 13, 82–83. [Google Scholar]
- LaFon, S.W.; Nelson, D.J. Purine metabolism in the intact sporozoites and merozoites of Eimeria tenella. Mol. Biochem. Parasitol. 1985, 14, 11–22. [Google Scholar] [CrossRef]
- Inaoka, D.K.; Iida, M.; Hashimoto, S.; Tabuchi, T.; Kuranaga, T.; Balogun, E.O.; Honma, T.; Tanaka, A.; Harada, S.; Nara, T.; et al. Design and synthesis of potent substrate-based inhibitors of the Trypanosoma cruzi dihydroorotate dehydrogenase. Bioorg. Med. Chem. 2017, 25, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Inaoka, D.K.; Iida, M.; Tabuchi, T.; Honma, T.; Lee, N.; Hashimoto, S.; Matsuoka, S.; Kuranaga, T.; Sato, K.; Shiba, T.; et al. The open form inducer approach for structure-based drug design. PLoS ONE 2016, 11, e0167078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohishi, T.; Masuda, T.; Abe, H.; Hayashi, C.; Adachi, H.; Ohba, S.-I.; Igarashi, M.; Watanabe, T.; Mimuro, H.; Amalia, E.; et al. Monotherapy with a novel intervenolin derivative, AS-1934, is an effective treatment for Helicobacter pylori infection. Helicobacter 2018, 23, e12470. [Google Scholar] [CrossRef]
- Pramisandi, A.; Dobashi, K.; Mori, M.; Nonaka, K.; Matsumoto, A.; Tokiwa, T.; Higo, M.; Kristiningrum; Amalia, E.; Nurkanto, A.; et al. Microbial inhibitors active against Plasmodium falciparum dihydroorotate dehydrogenase derived from an Indonesian soil fungus, Talaromyces pinophilus BioMCC-f.T.3979. J. Gen. Appl. Microbiol. 2020. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ghate, M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev. Med. Chem. 2011, 11, 1039–1055. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Awakawa, T.; Matsuzaki, M.; Cho, R.; Matsuda, Y.; Hoshino, S.; Shinohara, Y.; Yamamoto, M.; Kido, Y.; Inaoka, D.K.; et al. Complete biosynthetic pathways of ascofuranone and ascochlorin in Acremonium egyptiacum. Proc. Natl. Acad. Sci. USA 2019, 116, 8269–8274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanos-Cuentas, A.; Casapia, M.; Chuquiyauri, R.; Hinojosa, J.C.; Kerr, N.; Rosario, M.; Toovey, S.; Arch, R.H.; Phillips, M.A.; Rozenberg, F.D.; et al. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018, 18, 874–883. [Google Scholar] [CrossRef]
- Collins, K.A.; Rückle, T.; Elliott, S.; Marquart, L.; Ballard, E.; Chalon, S.; Griffin, P.; Möhrle, J.J.; McCarthy, J.S. DSM265 at 400 Milligrams Clears Asexual Stage Parasites but Not Mature Gametocytes from the Blood of Healthy Subjects Experimentally Infected with Plasmodium falciparum. Antimicrob. Agents Chemother. 2019, 63, e01837-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulyok, M.; Ruckle, T.; Roth, A.; Murbeth, R.E.; Chalon, S.; Kerr, N.; Samec, S.S.; Gobeau, N.; Calle, C.L.; Ibanez, J.; et al. DSM265 for Plasmodium falciparum chemoprophylaxis: A randomised, double blinded, phase 1 trial with controlled human malaria infection. Lancet Infect. Dis. 2017, 17, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Neidhardt, E.A.; Grossman, T.H.; Ocain, T.; Clardy, J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Kokkonda, S.; El Mazouni, F.; White, J.; Burrows, J.N.; Kaminsky, W.; Charman, S.A.; Matthews, D.; Rathod, P.K.; Phillips, M.A. Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 2014, 57, 5381–5394. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Le Nours, J.; Johansson, E.; Antal, T.; Ullrich, A.; Loffler, M.; Larsen, S. Inhibitor binding in a class 2 dihydroorotate dehydrogenase causes variations in the membrane-associated N-terminal domain. Protein Sci. 2004, 13, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Norager, S.; Jensen, K.F.; Bjornberg, O.; Larsen, S.E. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure 2002, 10, 1211–1223. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gao, Z.Q.; Liu, C.P.; Xu, J.H.; Li, L.F.; Ji, C.N.; Su, X.D.; Dong, Y.H. Structure of the putative dihydroorotate dehydrogenase from Streptococcus mutans. Acta Cryst. Sect. F Struct Biol. Cryst. Commun. 2011, 67, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Arakaki, T.L.; Buckner, F.S.; Gillespie, J.R.; Malmquist, N.A.; Phillips, M.A.; Kalyuzhniy, O.; Luft, J.R.; DeTitta, G.T.; Verlinde, C.L.M.J.; Van Voorhis, W.C.; et al. Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol. Microbiol. 2008, 68, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Tani, O.; Yamaguchi, T.; Namatame, I.; Sakashita, H.; Furukawa, K.; Yamasaki, K. Crystal structures of FMN-bound and FMN-free forms of dihydroorotate dehydrogenase from Trypanosoma brucei. FEBS Open Bio 2018, 8, 680–691. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Gujjar, R.; El Mazouni, F.; Kaminsky, W.; Malmquist, N.A.; Goldsmith, E.J.; Rathod, P.K.; Phillips, M.A. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Matsubayashi, M.; Hatta, T.; Miyoshi, T.; Anisuzzaman; Sasai, K.; Shimura, K.; Isobe, T.; Kita, K.; Tsuji, N. High-throughput RNA sequencing profiles and transcriptional evidence of aerobic respiratory enzymes in sporulating oocysts and sporozoites of Eimeria tenella. Infect. Genet. Evol. 2013, 18, 269–276. [Google Scholar] [CrossRef]
- Hartree, E.F. Determination of protein: A modification of the lowry method that gives a linear photometric response. Anal. Biochem. 1972, 48, 422–427. [Google Scholar] [CrossRef]
- Hartuti, E.D.; Inaoka, D.K.; Komatsuya, K.; Miyazaki, Y.; Miller, R.J.; Xinying, W.; Sadikin, M.; Prabandari, E.E.; Waluyo, D.; Kuroda, M.; et al. Biochemical studies of membrane bound Plasmodium falciparum mitochondrial L-malate:quinone oxidoreductase, a potential drug target. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Miyazaki, Y.; Inaoka, D.K.; Hartuti, E.D.; Watanabe, Y.I.; Shiba, T.; Harada, S.; Saimoto, H.; Burrows, J.N.; Benito, F.J.G.; et al. Identification of Plasmodium falciparum Mitochondrial Malate: Quinone Oxidoreductase Inhibitors from the Pathogen Box. Genes 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzym. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. D Biol Cryst. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. D Biol. Cryst. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Shiba, T.; Inaoka, D.K.; Takahashi, G.; Tsuge, C.; Kido, Y.; Young, L.; Ueda, S.; Balogun, E.O.; Nara, T.; Honma, T.; et al. Insights into the ubiquinol/dioxygen binding and proton relay pathways of the alternative oxidase. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Iltzsch, M.H. Pyrimidine salvage pathways in Toxoplasma gondii. J. Eukaryot. Microbiol. 1993, 40, 24–28. [Google Scholar] [CrossRef]
- Cassera, M.B.; Zhang, Y.; Hazleton, K.Z.; Schramm, V.L. Purine and pyrimidine pathways as targets in Plasmodium falciparum. Curr. Top. Med. Chem. 2011, 11, 2103–2115. [Google Scholar] [CrossRef]
- Copeland, R.A.; Marcinkeviciene, J.; Haque, T.S.; Kopcho, L.M.; Jiang, W.; Wang, K.; Ecret, L.D.; Sizemore, C.; Amsler, K.A.; Foster, L.; et al. Helicobacter pylori-selective antibacterials based on inhibition of pyrimidine biosynthesis. J. Biol. Chem. 2000, 275, 33373–33378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Moreno, G.; Sánchez-Carrasco, P.; Rúiz-Pérez, L.; Johansson, N.; Müller, S.; Baragaña, B.; Hampton, S.; Gilbert, I.; Kaiser, M.; Sarkar, S.; et al. Validation of Plasmodium falciparum dUTPase as the target of 52-tritylated deoxyuridine analogues with anti-malarial activity. Malar. J. 2019, 18, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, S.E.; Baragaña, B.; Schipani, A.; Bosch-Navarrete, C.; Musso-Buendía, J.A.; Recio, E.; Kaiser, M.; Whittingham, J.L.; Roberts, S.M.; Shevtsov, M.; et al. Design, Synthesis, and Evaluation of 5′-Diphenyl Nucleoside Analogues as Inhibitors of the Plasmodium falciparum dUTPase. ChemMedChem 2011, 6, 1816–1831. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kehrer, J.; Singer, M.; Reinig, M.; Santos, J.M.; Mair, G.R.; Frischknecht, F. Functional genetic evaluation of DNA house-cleaning enzymes in the malaria parasite: dUTPase and Ap4AH are essential in Plasmodium berghei but ITPase and NDH are dispensable. Expert Opin. Ther. Targets 2019, 23, 251–261. [Google Scholar] [CrossRef]
- Matsubayashi, M.; Inaoka, D.K.; Komatsuya, K.; Hatta, T.; Kawahara, F.; Sakamoto, K.; Hikosaka, K.; Yamagishi, J.; Sasai, K.; Shiba, T.; et al. Novel Characteristics of Mitochondrial Electron Transport Chain from Eimeria tenella. Genes 2019, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, J.; Farajallah, A.M.; Malmquist, N.A.; Rathod, P.K.; Phillips, M.A. Malarial dihydroorotate dehydrogenase. Substrate and inhibitor specificity. J. Biol. Chem. 2002, 277, 41827–41834. [Google Scholar] [CrossRef] [Green Version]
- Hurt, D.E.; Widom, J.; Clardy, J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Cryst. D Biol. Cryst. 2006, 62, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Lahouel, M.; Zini, R.; Zellagui, A.; Rhouati, S.; Carrupt, P.-A.; Morin, D. Ferulenol specifically inhibits succinate ubiquinone reductase at the level of the ubiquinone cycle. Biochem. Biophys. Res. Commun. 2007, 355, 252–257. [Google Scholar] [CrossRef]
- Gebauer, M. Synthesis and structure-activity relationships of novel warfarin derivatives. Bioorg. Med. Chem. 2007, 15, 2414–2420. [Google Scholar] [CrossRef]
- Li, W.; Schulman, S.; Dutton, R.J.; Boyd, D.; Beckwith, J.; Rapoport, T.A. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 2010, 463, 507–512. [Google Scholar] [CrossRef]
- Chen, S.F.; Perrella, F.W.; Behrens, D.L.; Papp, L.M. Inhibition of dihydroorotate dehydrogenase activity by brequinar sodium. Cancer Res. 1992, 52, 3521–3527. [Google Scholar] [PubMed]
- Phillips, M.A.; Rathod, P.K. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Disord Drug Targets 2010, 10, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, D.; Hartuti, E.D.; Inaoka, D.K.; Sakura, T.; Amalia, E.; Nagahama, M.; Yoshioka, Y.; Tsuji, N.; Nozaki, T.; Kita, K.; et al. Structural and Biochemical Features of Eimeria tenella Dihydroorotate Dehydrogenase, a Potential Drug Target. Genes 2020, 11, 1468. https://doi.org/10.3390/genes11121468
Sato D, Hartuti ED, Inaoka DK, Sakura T, Amalia E, Nagahama M, Yoshioka Y, Tsuji N, Nozaki T, Kita K, et al. Structural and Biochemical Features of Eimeria tenella Dihydroorotate Dehydrogenase, a Potential Drug Target. Genes. 2020; 11(12):1468. https://doi.org/10.3390/genes11121468
Chicago/Turabian StyleSato, Dan, Endah Dwi Hartuti, Daniel Ken Inaoka, Takaya Sakura, Eri Amalia, Madoka Nagahama, Yukina Yoshioka, Naotoshi Tsuji, Tomoyoshi Nozaki, Kiyoshi Kita, and et al. 2020. "Structural and Biochemical Features of Eimeria tenella Dihydroorotate Dehydrogenase, a Potential Drug Target" Genes 11, no. 12: 1468. https://doi.org/10.3390/genes11121468
APA StyleSato, D., Hartuti, E. D., Inaoka, D. K., Sakura, T., Amalia, E., Nagahama, M., Yoshioka, Y., Tsuji, N., Nozaki, T., Kita, K., Harada, S., Matsubayashi, M., & Shiba, T. (2020). Structural and Biochemical Features of Eimeria tenella Dihydroorotate Dehydrogenase, a Potential Drug Target. Genes, 11(12), 1468. https://doi.org/10.3390/genes11121468