Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas


Abstract
:1. Introduction
2. Materials and Methods
3. Results
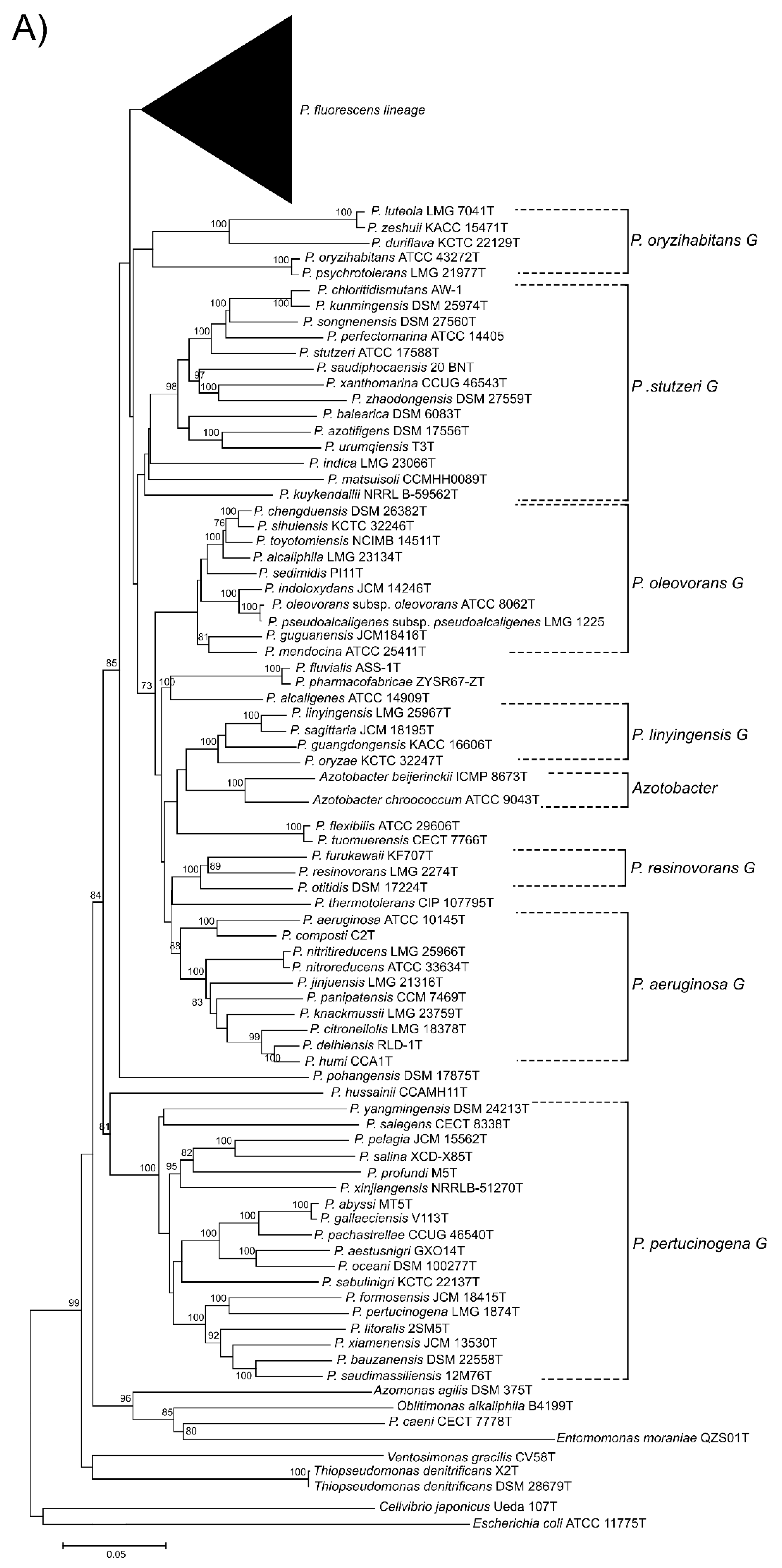
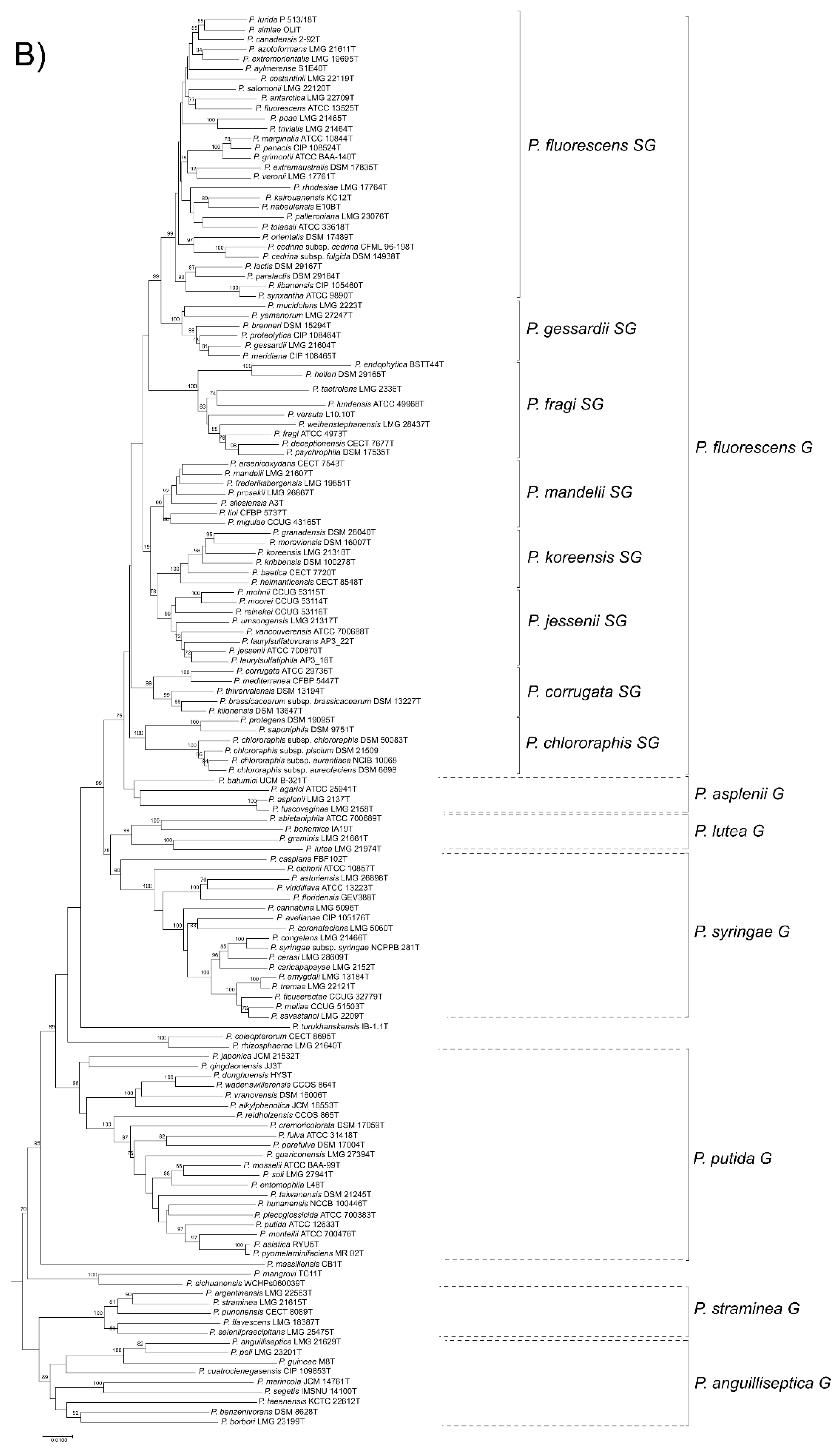
3.1. 16S rDNA Phylogeny
3.2. Four-Gene MLSA
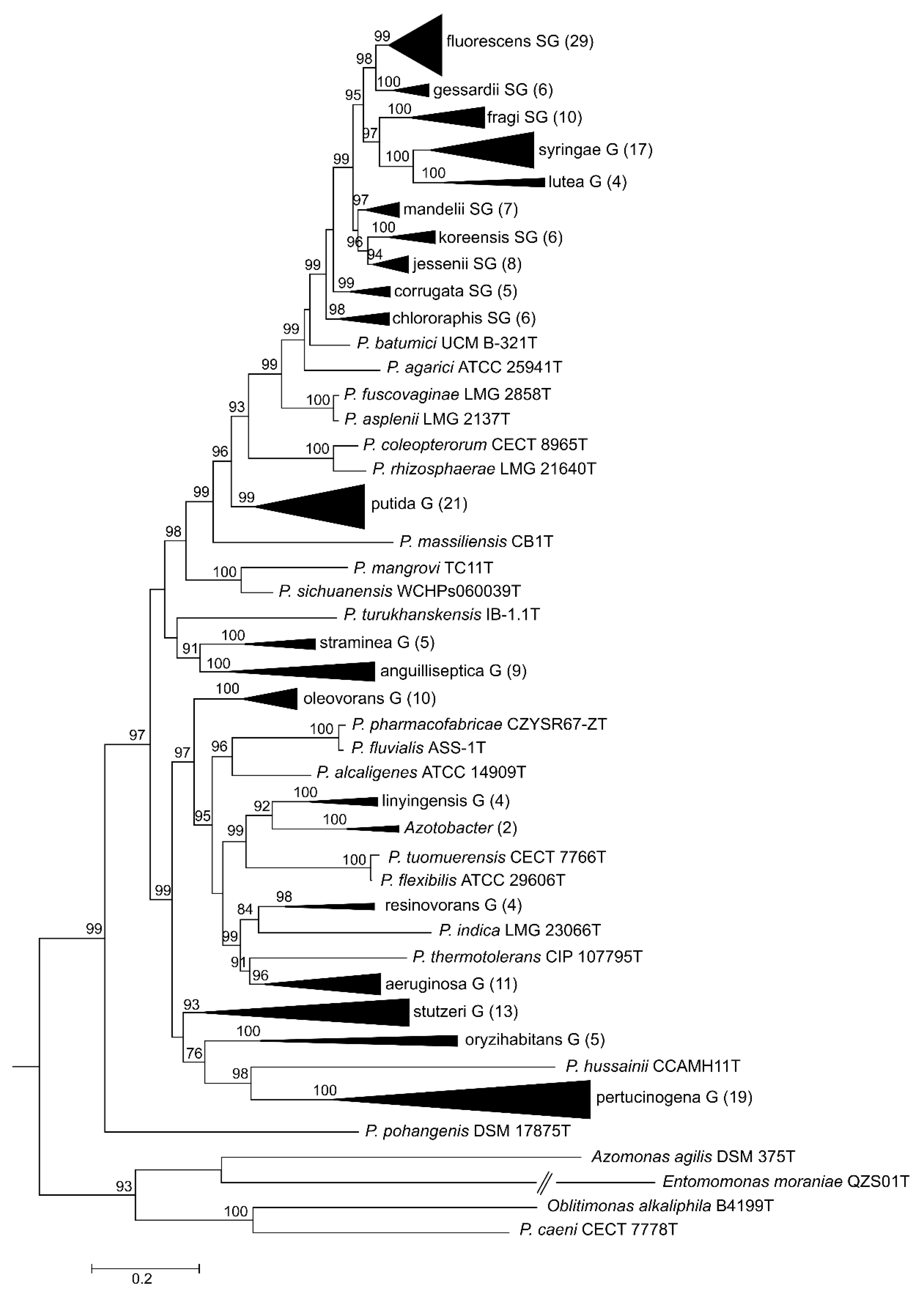
3.3. Phylogeny Based on 100 Gene Sequences
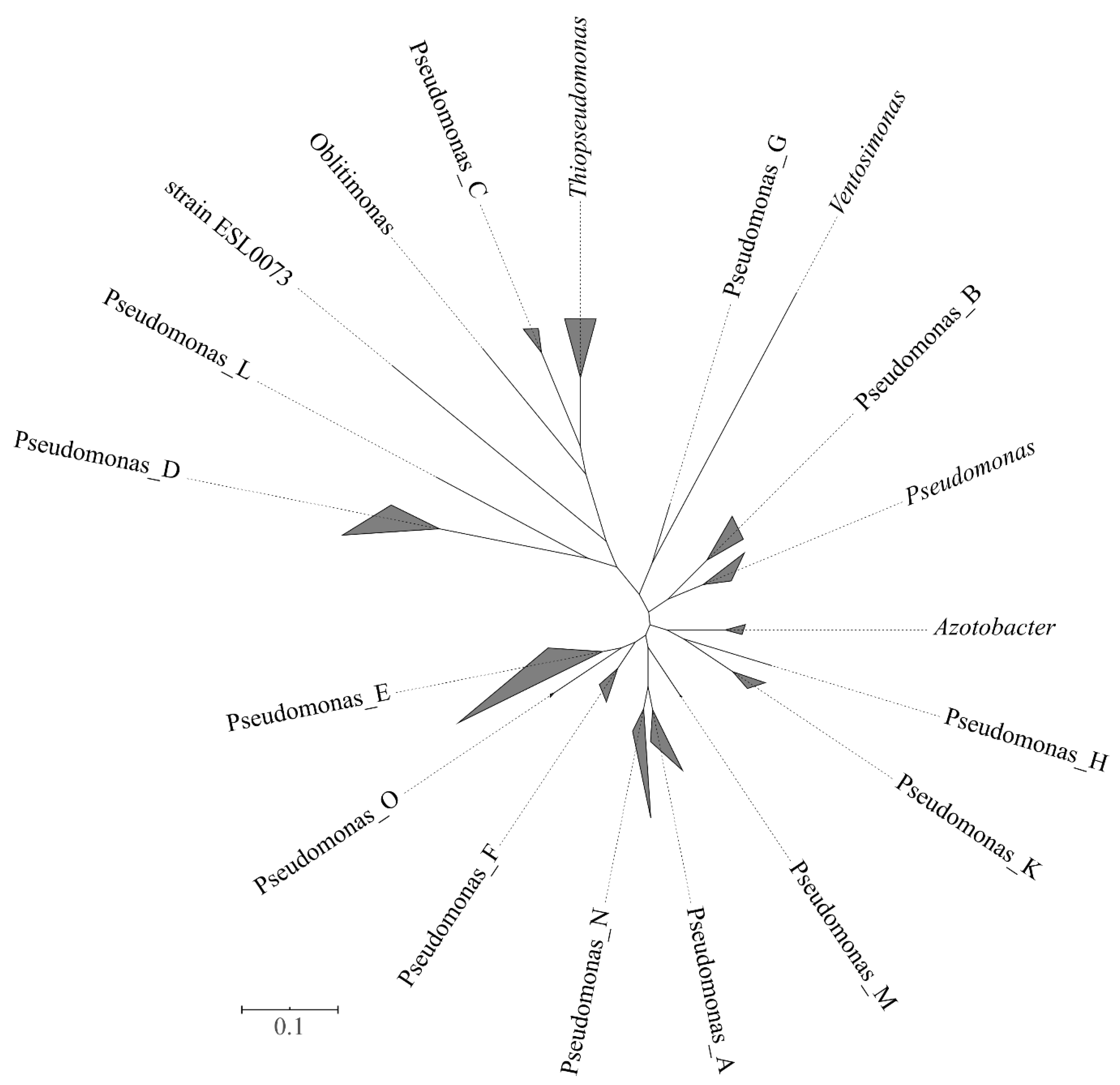
3.4. Analyses Performed at the Genome Taxonomy Database (GTDB Taxonomy)
3.5. Analysis of the Percentage of Conserved Proteins (POCP)
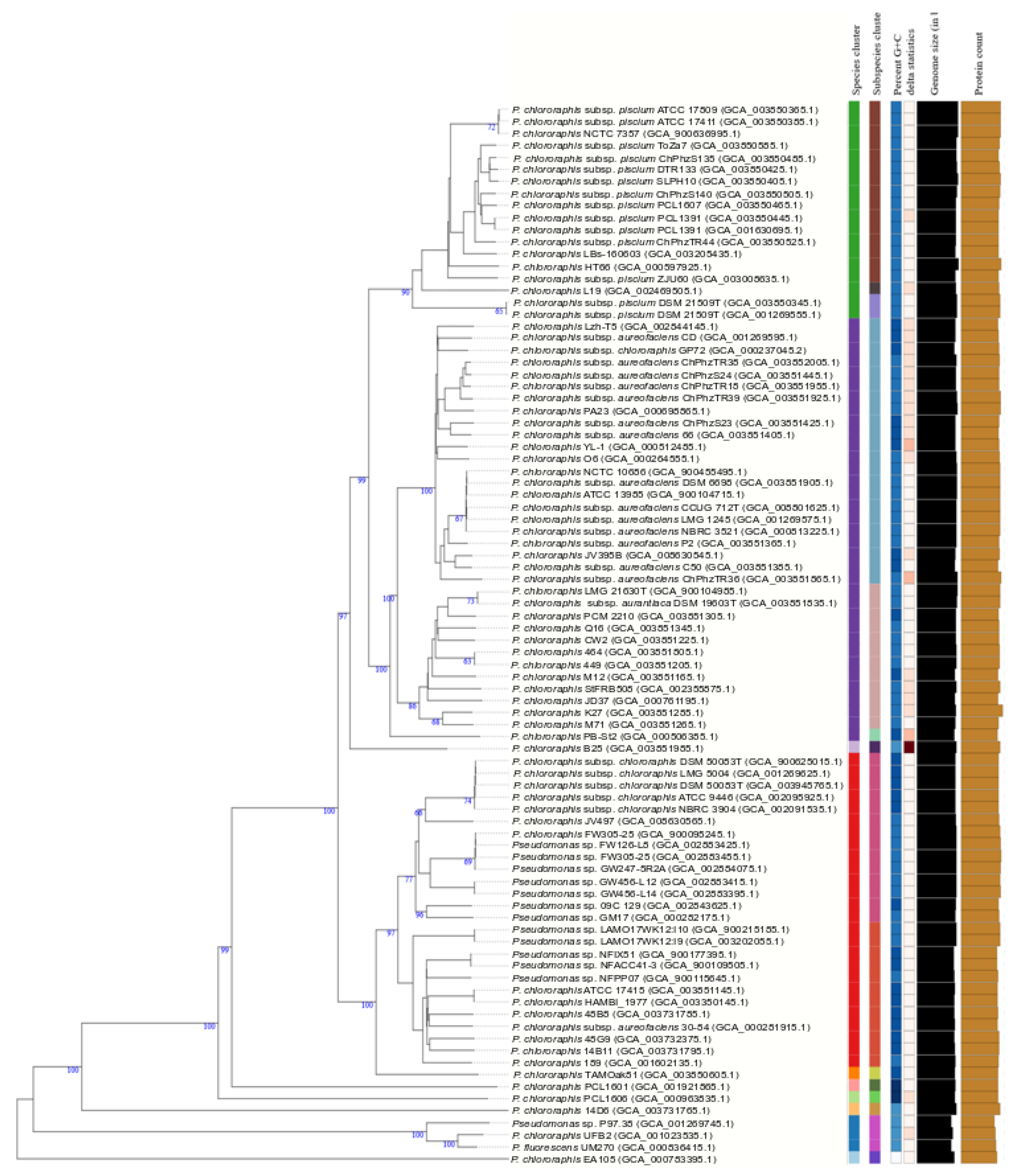
3.6. Pseudomonas Chlororaphis Case Study
4. Discussion
4.1. Species and Subspecies Delineation
4.2. Genus Delineation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [Green Version]
- Mulet, M.; Lalucat, J.; García-Valdés, E. DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 2010, 12, 1513–1530. [Google Scholar]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.D.; Kämpfer, P.; Maiden, M.C.J.; Nesme, X.; Rossello-Mora, R.; Swings, J.; Trüper, H.G.; et al. Report of the ad hoc committee for there-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002, 52, 1043–1047. [Google Scholar] [CrossRef]
- Maiden, M.C.J.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotech. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Parte, A.C. LPSN—List of Prokaryotic names with Standing in Nomenclature (bacterio.net), 20 years on. Int. J. Syst. Evol. Microbiol. 2018, 68, 1825–1829. [Google Scholar] [CrossRef]
- Palleroni, N.J. Genus I Pseudomonas. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Brenner, D.J., Krieg, N.R., Staley, J.T., Garrity, G.M., Eds.; Springer: East Lansing, MI, USA, 2005; Volume 2, pp. 323–379. [Google Scholar] [CrossRef]
- Gomila, M.; Peña, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. Phylogenomics and systematics in Pseudomonas. Front. Microbial. 2015, 6, 214. [Google Scholar] [CrossRef] [Green Version]
- Peix, A.; Ramírez-Bahena, M.H.; Velázquez, E. The current status on the taxonomy of Pseudomonas revisited: An update. Infect. Genet. Evol. 2018, 57, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gomila, M.; Busquets, A.; Mulet, M.; García-Valdés, E.; Lalucat, J. Clarification of taxonomic status within the Pseudomonas syringae species group based on a phylogenomic analysis. Front. Microbiol. 2017, 8, 2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Sanz, D.; Meier-Kolthoff, J.P.; Göker, M.; Martin, M.; Rivilla, R.; Redondo-Nieto, M. Genomic and genetic diversity within the Pseudomonas fluorescens complex. PLoS ONE 2016, 11, e0150183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Carranza, E.; Ponce-Soto, G.Y.; Servín-González, L.; Alcaraz, L.D.; Soberón-Chávez, G. Evolution of bacteria seen through their essential genes: The case of Pseudomonas aeruginosa and Azotobacter vinelandii. Microbiology 2019, 165, 9. [Google Scholar] [CrossRef] [PubMed]
- Peña, A.; Busquets, A.; Gomila, M.; Mulet, M.; Gomila, R.M.; Garcia-Valdes, E.; Reddy, T.B.K.; Huntemann, M.; Varghese, N.; Ivanova, N.; et al. High-quality draft genome sequences of Pseudomonas monteilii DSM 14164T, Pseudomonas mosselii DSM 17497T, Pseudomonas plecoglossicida DSM 15088T, Pseudomonas taiwanensis DSM 21245T and Pseudomonas vranovensis DSM 16006T: Taxonomic considerations. Access Microbiol. 2019, 1, e000067. [Google Scholar] [CrossRef]
- Hesse, C.; Schulz, F.; Bull, C.T.; Shaffer, B.T.; Yan, Q.; Shapiro, N.; Hassan, K.A.; Varghese, N.; Elbourne, L.D.H.; Paulsen, I.T.; et al. Genome-based evolutionary history of Pseudomonas spp. Environ. Microbiol. 2018, 20, 2142–2159. [Google Scholar] [CrossRef]
- Kyrpides, N.C.; Woyke, T.; Eisen, J.A.; Garrity, G.; Lilburn, T.G.; Beck, B.J.; Whitman, W.B.; Hugenholtz, P.; Klenk, H.P. Genomic encyclopedia of type strains, phase I: The one thousand microbial genomes (KMG-I) project. Stand. Genom. Sci. 2014, 9, 1278–1284. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Mulet, M.; Gomila, M.; Ramírez, A.; Cardew, S.; Moore, E.R.B.; Lalucat, J.; García-Valdés, E. Uncommonly isolated clinical Pseudomonas: Identification and phylogenetic assignation. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 351–359. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- JSpeciesWS. JSpeciesWS Home Page. Available online: http://jspecies.ribohost.com/jspeciesws/ (accessed on 20 December 2019).
- GTDB. Genome Taxonomy Database. Available online: http://gtdb.ecogenomic.org/ (accessed on 30 September 2019).
- Meier-Kolthoff, J.P.; Göcker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Type Strain Genome Server (TYGS) Platform. Available online: https://tygs.dsmz.de/ (accessed on 10 December 2019).
- Qin, Q.L.; Xie, B.B.; Zhang, X.Y.; Chen, X.L.; Zhou, B.C.; Zhou, J.; Oren, A.; Zhang, Y.Z. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef] [Green Version]
- IGM. Phylogenetic Profiler for Single Genes Tool. Available online: https://img.jgi.doe.gov/ (accessed on 15 November 2019).
- Cladera, A.M.; García-Valdés, E.; Lalucat, J. Genotype versus phenotype in the circumscription of bacterial species: The case of Pseudomonas stutzeri and Pseudomonas chloritidismutans. Arch. Microbiol. 2006, 184, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–667. [Google Scholar] [CrossRef]
- Yumoto, I.; Kusano, T.; Shingyo, T.; Nodasaka, Y.; Matsuyama, H.; Okuyama, H. Assignment of Pseudomonas sp. strain E-3 to Pseudomonas psychrophila sp. nov., a new facultatively psychrophilic bacterium. Extremophiles 2001, 5, 343–349. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kasai, H.; Arnold, D.L.; Jackson, R.W.; Vivian, A.; Harayama, S. Phylogeny of the genus Pseudomonas: Intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 2000, 146, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Tayeb, L.A.; Ageron, E.; Grimont, F.; Grimont, P.A.D. Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates. Res. Microbiol. 2005, 156, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Skerman, V.B.D.; McGowan, V.; Sneath, P.H.A. Approved lists of bacterial names. Int. J. Syst. Bacteriol. 1980, 30, 225–420. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Palleroni, N. Deoxyribonucleic acid similarities among Pseudomonas species. Int. J. Syst. Bacteriol. 1989, 39, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Peix, A.; Valverde, A.; Rivas, R.; Igual, J.M.; Ramírez-Bahena, M.; Mateos, P.F.; Santa-Regina, I.; Rodríguez-Barrueco, C.; Martínez-Molina, E.; Velázquez, E. Reclassification of Pseudomonas aurantiaca as a synonym of Pseudomonas chlororaphis and proposal of three subspecies, P. chlororaphis subsp. chlororaphis subsp. nov., P. chlororaphis subsp. aureofaciens subsp. nov., comb. nov. and P. chlororaphis subsp. aurantiaca subsp. nov., comb. nov. Int. J. System. Evol. Microbiol. 2007, 57, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Burr, S.E.; Gobeli, S.; Kuhnert, P.; Goldschmidt-Clermont, E.; Frey, J. Pseudomonas chlororaphis subsp. piscium subsp. nov., isolated from freshwater fish. Int. J. System. Evol. Microbiol. 2010, 60, 2753–2757. [Google Scholar] [CrossRef] [Green Version]
- Mende, D.R.; Sunagawa, S.; Zeller, G.; Bork, P. Accurate and universal delineation of prokaryotic species. Nat. Methods 2013, 10, 881–884. [Google Scholar] [CrossRef]
- Rossello-Mora, R.; Amann, R. Past and future species definitions for Bacteria and Archaea. System. Appl. Microbiol. 2015, 38, 209–216. [Google Scholar] [CrossRef]
- Beye, M.; Fahsi, N.; Raoult, D.; Fournier, P.-E. Careful use of 16S rRNA gene sequence similarity values for the identification of Mycobacterium species. New Microbes New Infect. 2018, 22, 24–29. [Google Scholar] [CrossRef]
- Rediers, H.; Vanderleyden, J.; De Mot, R. Azotobacter vinelandii: A Pseudomonas in disguise? Microbiology 2004, 150, 1117–1119. [Google Scholar] [CrossRef]
- Özen, A.I.; Ussery, D.W. Defining the Pseudomonas genus: Where do we draw the line with Azotobacter? Microb. Ecol. 2012, 63, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.G.; Kukavica-Ibrulj, I.; Dupont, M.J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol. Evol. 2018, 11, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanier, R.Y.; Ingraham, J.L.; Wheelis, M.L.; Paintes, P.R. The Microbial World, 5th ed.; Prentice-Hall: Englewood Cliffs, NJ, USA, 1986. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synonyms | ANIb a | 4-gene MLSA | Reclassification |
|---|---|---|---|
| P. asplenii | 97.91 | 99.6 | P. aspleniii |
| P. fuscovaginae | |||
| P. meliae | >97 | 98.2 | P. amygdali |
| P. amygdali | 100 | ||
| P. savastanoi | 98.3 | ||
| P. ficuserectae | 98.6 | ||
| P. asiatica | 99.91 | 100 | P. asiatica |
| P. pyomelaninifaciens | |||
| P. chloritidismutans | 96.29 | 99.8 | ‘P. chloritidismutans’ b |
| P. kunmingensis | |||
| P. oleovorans subsp. oleovorans | 96.75 | 99.9 | P. oleovorans |
| P. indoloxidans | |||
| P. flexibilis | 100 | 99.5 | P. flexibilis |
| P. tuomuerensis | |||
| P. fluvialis | 98.46 | 99.2 | P. fluvialis |
| P. pharmacofabricae | |||
| P. nitritireducens | - | 99.4 | P. nitroreducens |
| P. nitroreducens | |||
| P. citronellolis | 95.9 | 99.5 | P. citronellolis |
| P. humi | |||
| P. oryzihabitans | 97.70 | 99.7 | P. oryzihabitans |
| P. psychrotolerans | |||
| P. luteola | 97.60 | 99.3 | P. luteola |
| P. zeshuii | |||
| P. abyssi | 97.10 | 99.7 | P. gallaeciensis |
| P. gallaeciensis |
| Proposed Genera and Species in the GTDB Taxonomy | Accepted Taxonomy and Phylogenetic Groups | ||
|---|---|---|---|
| Genera | nr. species | Genus, group (G) or representative species | nr. species |
| Azotobacter | 3 | Azotobacter | 8 |
| Oblitimonas | 1 | O. alcaliphila | 1 |
| Pseudomonas | 14 | aeruginosa G | 10 |
| Pseudomonas_A | 38 | stutzeri G | 12 |
| Pseudomonas_B | 7 | oryzihabitans G | 5 |
| Pseudomonas_C | 3 | P. caeni | 1 |
| Pseudomonas_D | 18 | pertucinogena G | 18 |
| Pseudomonas_E | 404 | anguiliseptica G | 142 |
| fluorescens G | |||
| lutea G | |||
| putida G | |||
| oleovorans G | |||
| straminea G | |||
| syringae G | |||
| Pseudomonas_F | 9 | P. resinovorans | 4 |
| Pseudomonas_G | 1 | P. thermotolerans | 1 |
| Pseudomonas_H | 1 | P. flexibilis | 1 |
| Pseudomonas_K | 4 | linyingensis G | 4 |
| Pseudomonas_L | 1 | P. hussainii | 1 |
| Pseudomonas_M | 2 | P. indica | 1 |
| Pseudomonas_N | 2 | P. azotifigens | 1 |
| Pseudomonas_O | 2 | P. kuykendallii | 1 |
| Thiopseudomonas | 2 | T. denitrificans | 1 |
| strain ESL0073 | 1 | - | 0 |
| Ventosimonas | 1 | V. gracilis | 1 |
| Total number of species | 514 | 213 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalucat, J.; Mulet, M.; Gomila, M.; García-Valdés, E. Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas. Genes 2020, 11, 139. https://doi.org/10.3390/genes11020139
Lalucat J, Mulet M, Gomila M, García-Valdés E. Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas. Genes. 2020; 11(2):139. https://doi.org/10.3390/genes11020139
Chicago/Turabian StyleLalucat, Jorge, Magdalena Mulet, Margarita Gomila, and Elena García-Valdés. 2020. "Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas" Genes 11, no. 2: 139. https://doi.org/10.3390/genes11020139
APA StyleLalucat, J., Mulet, M., Gomila, M., & García-Valdés, E. (2020). Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas. Genes, 11(2), 139. https://doi.org/10.3390/genes11020139