Working on Genomic Stability: From the S-Phase to Mitosis


Abstract
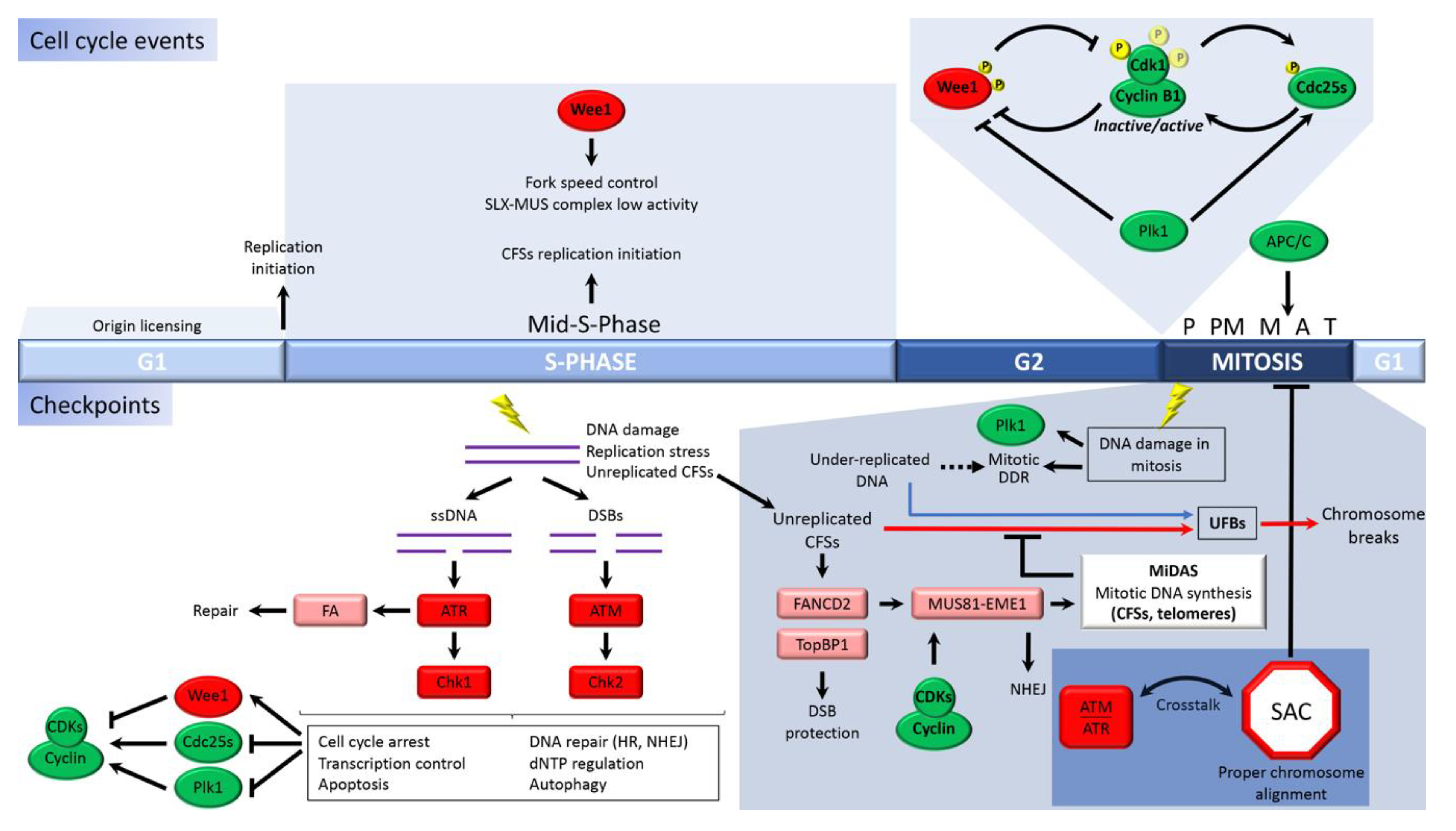
:1. Introduction
2. Replication Stress and Under-Replicated DNA in Mitosis
2.1. Common Fragile Sites
2.2. Fallible Checkpoints Lead to Premature Mitosis with Under-Replicated DNA
3. Crosstalk between the S-Phase and Mitosis
3.1. DNA Synthesis of Under-Replicated DNA Tracks Prior to Mitosis
3.2. Mitotic DNA Synthesis
4. DNA Damage in Mitosis
4.1. Mitotic DDR
4.2. Repair in Mitosis
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Widrow, R.J.; Hansen, R.S.; Kawame, H.; Gartler, S.M.; Laird, C.D. Very late DNA replication in the human cell cycle. Proc. Natl. Acad. Sci. USA 1998, 95, 11246–11250. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Diffley, J.F. Regulation of early events in chromosome replication. Curr. Biol. 2004, 14, R778–R786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akopyan, K.; Silva Cascales, H.; Hukasova, E.; Saurin, A.T.; Mullers, E.; Jaiswal, H.; Hollman, D.A.; Kops, G.J.; Medema, R.H.; Lindqvist, A. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol. Cell 2014, 53, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Lafarga, V.; Sung, H.M.; Haneke, K.; Roessig, L.; Pauleau, A.L.; Bruer, M.; Rodriguez-Acebes, S.; Lopez-Contreras, A.J.; Gruss, O.J.; Erhardt, S.; et al. TIAR marks nuclear G2/M transition granules and restricts CDK1 activity under replication stress. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Irony-Tur Sinai, M.; Kerem, B. DNA replication stress drives fragile site instability. Mutat. Res. 2018, 808, 56–61. [Google Scholar] [CrossRef]
- Crncec, A.; Hochegger, H. Triggering mitosis. FEBS Lett. 2019, 593, 2868–2888. [Google Scholar] [CrossRef] [Green Version]
- Hegarat, N.; Rata, S.; Hochegger, H. Bistability of mitotic entry and exit switches during open mitosis in mammalian cells. BioEssays 2016, 38, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, H.; Benada, J.; Mullers, E.; Akopyan, K.; Burdova, K.; Koolmeister, T.; Helleday, T.; Medema, R.H.; Macurek, L.; Lindqvist, A. ATM/Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terabayashi, T.; Hanada, K. Genome instability syndromes caused by impaired DNA repair and aberrant DNA damage responses. Cell Biol. Toxicol. 2018, 34, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Hayward, D.; Alfonso-Perez, T.; Gruneberg, U. Orchestration of the spindle assembly checkpoint by CDK1-cyclin B1. FEBS Lett. 2019, 593, 2889–2907. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, E.R.; Scheuringer, N.; Podtelejnikov, A.V.; Mann, M.; Peters, J.M. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 2000, 11, 1555–1569. [Google Scholar] [CrossRef] [Green Version]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Labib, K.; De Piccoli, G. Surviving chromosome replication: The many roles of the S-phase checkpoint pathway. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2011, 366, 3554–3561. [Google Scholar] [CrossRef] [Green Version]
- Blow, J.J.; Dutta, A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005, 6, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Diffley, J.F. Eukaryotic DNA replication control: Lock and load, then fire. Curr. Opin. Cell Biol. 2009, 21, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Musialek, M.W.; Rybaczek, D. Behavior of replication origins in Eukaryota—Spatio-temporal dynamics of licensing and firing. Cell Cycle 2015, 14, 2251–2264. [Google Scholar] [CrossRef] [PubMed]
- Limas, J.C.; Cook, J.G. Preparation for DNA replication: The key to a successful S phase. FEBS Lett. 2019, 593, 2853–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Ishimi, Y. Regulation of MCM2-7 function. Genes Genet. Syst. 2018, 93, 125–133. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Liu, P.; Slater, D.M.; Lenburg, M.; Nevis, K.; Cook, J.G.; Vaziri, C. Replication licensing promotes cyclin D1 expression and G1 progression in untransformed human cells. Cell Cycle 2009, 8, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Nevis, K.R.; Cordeiro-Stone, M.; Cook, J.G. Origin licensing and p53 status regulate Cdk2 activity during G(1). Cell Cycle 2009, 8, 1952–1963. [Google Scholar] [CrossRef] [Green Version]
- Shreeram, S.; Sparks, A.; Lane, D.P.; Blow, J.J. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene 2002, 21, 6624–6632. [Google Scholar] [CrossRef] [Green Version]
- Kunnev, D.; Rusiniak, M.E.; Kudla, A.; Freeland, A.; Cady, G.K.; Pruitt, S.C. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 2010, 29, 3630–3638. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, S.C.; Bailey, K.J.; Freeland, A. Reduced Mcm2 expression results in severe stem/progenitor cell deficiency and cancer. Stem Cells 2007, 25, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef]
- Matson, J.P.; House, A.M.; Grant, G.D.; Wu, H.; Perez, J.; Cook, J.G. Intrinsic checkpoint deficiency during cell cycle re-entry from quiescence. J. Cell Biol. 2019, 218, 2169–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, J.J.; Gillespie, P.J. Replication licensing and cancer–A fatal entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, L.W.; Lents, N.H.; Baldassare, J.J. Cyclin A-CDK activity during G1 phase impairs MCM chromatin loading and inhibits DNA synthesis in mammalian cells. Cell Cycle 2008, 7, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Diffley, J.F. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Petersen, B.O.; Lukas, J.; Sorensen, C.S.; Bartek, J.; Helin, K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999, 18, 396–410. [Google Scholar] [CrossRef]
- Liu, E.; Li, X.; Yan, F.; Zhao, Q.; Wu, X. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J. Biol. Chem. 2004, 279, 17283–17288. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, N.; Tatsumi, Y.; Tsurumi, T.; Matsukage, A.; Kiyono, T.; Nishitani, H.; Fujita, M. Cdt1 phosphorylation by cyclin A-dependent kinases negatively regulates its function without affecting geminin binding. J. Biol. Chem. 2004, 279, 19691–19697. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.P.; Labib, K. Chromosome Duplication in Saccharomyces cerevisiae. Genetics 2016, 203, 1027–1067. [Google Scholar] [CrossRef] [Green Version]
- DePamphilis, M.L. The ‘ORC cycle’: A novel pathway for regulating eukaryotic DNA replication. Gene 2003, 310, 1–15. [Google Scholar] [CrossRef]
- Masuda, T.; Mimura, S.; Takisawa, H. CDK- and Cdc45-dependent priming of the MCM complex on chromatin during S-phase in Xenopus egg extracts: Possible activation of MCM helicase by association with Cdc45. Genes Cells 2003, 8, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ishimi, Y.; Tanudji, M.; Lees, E. Human CDK2 inhibition modifies the dynamics of chromatin-bound minichromosome maintenance complex and replication protein A. Cell Cycle 2005, 4, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Takase, Y.; Komori, Y.; Hashimoto, Y.; Arata, T.; Kamimura, Y.; Araki, H.; Takisawa, H. A novel ring-like complex of Xenopus proteins essential for the initiation of DNA replication. Genes Dev. 2003, 17, 1141–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [Green Version]
- Moritani, M.; Ishimi, Y. Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J. Biochem. 2013, 154, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Dou, Q.P.; Fridovich-Keil, J.L.; Pardee, A.B. Inducible proteins binding to the murine thymidine kinase promoter in late G1/S phase. Proc. Natl. Acad. Sci. USA 1991, 88, 1157–1161. [Google Scholar] [CrossRef] [Green Version]
- Ezhevsky, S.A.; Ho, A.; Becker-Hapak, M.; Davis, P.K.; Dowdy, S.F. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol. Cell. Biol. 2001, 21, 4773–4784. [Google Scholar] [CrossRef] [Green Version]
- Masai, H.; Matsui, E.; You, Z.; Ishimi, Y.; Tamai, K.; Arai, K. Human Cdc7-related kinase complex. In vitro phosphorylation of MCM by concerted actions of Cdks and Cdc7 and that of a criticial threonine residue of Cdc7 bY Cdks. J. Biol. Chem. 2000, 275, 29042–29052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, M.; Rappas, M.; Wardlaw, C.P.; Garcia, V.; Ren, J.Y.; Day, M.; Carr, A.M.; Oliver, A.W.; Du, L.L.; Pearl, L.H. Phosphorylation-dependent assembly and coordination of the DNA damage checkpoint apparatus by Rad4(TopBP1). Mol. Cell 2013, 51, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Van Vugt, M.A.; Gardino, A.K.; Linding, R.; Ostheimer, G.J.; Reinhardt, H.C.; Ong, S.E.; Tan, C.S.; Miao, H.; Keezer, S.M.; Li, J.; et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010, 8, e1000287. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Burdova, K.; Lidak, T.; von Morgen, P.; Macurek, L. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015, 14, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orthwein, A.; Fradet-Turcotte, A.; Noordermeer, S.M.; Canny, M.D.; Brun, C.M.; Strecker, J.; Escribano-Diaz, C.; Durocher, D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014, 344, 189–193. [Google Scholar] [CrossRef]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol. Cell 2019, 73, 915–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.S.; Zhao, R.; Xu, X.; Ham, A.J.; Cortez, D. Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M checkpoint response to DNA damage. Cancer Res. 2007, 67, 6685–6690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupfer, G.M.; Yamashita, T.; Naf, D.; Suliman, A.; Asano, S.; D’Andrea, A.D. The Fanconi anemia polypeptide, FAC, binds to the cyclin-dependent kinase, cdc2. Blood 1997, 90, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Qiao, F.; Wilson, J.B.; High, A.A.; Schroeder, M.J.; Stukenberg, P.T.; Moss, A.; Shabanowitz, J.; Hunt, D.F.; Jones, N.J.; et al. FANCG is phosphorylated at serines 383 and 387 during mitosis. Mol. Cell. Biol. 2004, 24, 8576–8585. [Google Scholar] [CrossRef] [Green Version]
- St Onge, R.P.; Besley, B.D.; Pelley, J.L.; Davey, S. A role for the phosphorylation of hRad9 in checkpoint signaling. J. Biol. Chem. 2003, 278, 26620–26628. [Google Scholar] [CrossRef] [Green Version]
- Anantha, R.W.; Vassin, V.M.; Borowiec, J.A. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 2007, 282, 35910–35923. [Google Scholar] [CrossRef] [Green Version]
- Ira, G.; Malkova, A.; Liberi, G.; Foiani, M.; Haber, J.E. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 2003, 115, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 2006, 314, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, N.; Mukherjee, B.; Catherine Hardebeck, M.; Ilcheva, M.; Vanessa Camacho, C.; Louise Harris, J.; Porteus, M.; Llorente, B.; Khanna, K.K.; Burma, S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun. 2014, 5, 3561. [Google Scholar] [CrossRef]
- Luscher-Firzlaff, J.M.; Lilischkis, R.; Luscher, B. Regulation of the transcription factor FOXM1c by Cyclin E/CDK2. FEBS Lett. 2006, 580, 1716–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, E.M.; Haas, W.; Gygi, M.; Gygi, S.P.; Kellogg, D.R. Cdc28-dependent regulation of the Cdc5/Polo kinase. Curr. Biol. 2005, 15, 2033–2037. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Mueller, P.R.; Coleman, T.R.; Kumagai, A.; Dunphy, W.G. Myt1: A membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 1995, 270, 86–90. [Google Scholar] [CrossRef]
- Nilsson, I.; Hoffmann, I. Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 2000, 4, 107–114. [Google Scholar] [CrossRef]
- Ji, Z.; Gao, H.; Jia, L.; Li, B.; Yu, H. A sequential multi-target Mps1 phosphorylation cascade promotes spindle checkpoint signaling. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Morin, V.; Prieto, S.; Melines, S.; Hem, S.; Rossignol, M.; Lorca, T.; Espeut, J.; Morin, N.; Abrieu, A. CDK-dependent potentiation of MPS1 kinase activity is essential to the mitotic checkpoint. Curr. Biol. 2012, 22, 289–295. [Google Scholar] [CrossRef]
- Yudkovsky, Y.; Shteinberg, M.; Listovsky, T.; Brandeis, M.; Hershko, A. Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem. Biophys. Res. Commun. 2000, 271, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005, 434, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Shamanna, R.A.; de Freitas, J.K.; Okur, M.; Khadka, P.; Kulikowicz, T.; Holland, P.P.; Tian, J.; Croteau, D.L.; Davis, A.J.; et al. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017, 8, 2039. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Chen, J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [Green Version]
- Caspari, T.; Murray, J.M.; Carr, A.M. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 2002, 16, 1195–1208. [Google Scholar] [CrossRef] [Green Version]
- Terasawa, M.; Shinohara, A.; Shinohara, M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014, 10, e1004563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Huhn, D.; et al. RECQ5 Helicase Cooperates with MUS81 Endonuclease in Processing Stalled Replication Forks at Common Fragile Sites during Mitosis. Mol. Cell 2017, 66, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Matos, J.; Blanco, M.G.; Maslen, S.; Skehel, J.M.; West, S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011, 147, 158–172. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, H.D.; Sarbajna, S.; Matos, J.; West, S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 2013, 52, 234–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.W.; West, S.C. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat. Commun. 2014, 5, 4844. [Google Scholar] [CrossRef]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfander, B.; Diffley, J.F. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 2011, 30, 4897–4907. [Google Scholar] [CrossRef] [Green Version]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Fragkos, M.; Naim, V. Rescue from replication stress during mitosis. Cell Cycle 2017, 16, 613–633. [Google Scholar] [CrossRef] [Green Version]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2019, 43, e20190138. [Google Scholar] [CrossRef]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.C.; Cary, R.B.; Lakin, N.D.; Hann, B.C.; Teo, S.H.; Chen, D.J.; Jackson, S.P. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc. Natl. Acad. Sci. USA 1999, 96, 11134–11139. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Eychenie, S.; Decaillet, C.; Basbous, J.; Constantinou, A. DNA structure-specific priming of ATR activation by DNA-PKcs. J. Cell Biol. 2013, 202, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Z.; Bailis, J.M.; Johnson, S.A.; Dilworth, S.M.; Hunter, T. Rapid activation of ATM on DNA flanking double-strand breaks. Nat. Cell Biol. 2007, 9, 1311–1318. [Google Scholar] [CrossRef]
- Cuadrado, M.; Martinez-Pastor, B.; Fernandez-Capetillo, O. ATR activation in response to ionizing radiation: Still ATM territory. Cell Div. 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef] [Green Version]
- Ozeri-Galai, E.; Schwartz, M.; Rahat, A.; Kerem, B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 2008, 27, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Ataxia-telangiectasia mutated (ATM)-dependent activation of ATR occurs through phosphorylation of TopBP1 by ATM. J. Biol. Chem. 2007, 282, 17501–17506. [Google Scholar] [CrossRef] [Green Version]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, D.; Glick, G.; Elledge, S.J. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. USA 2004, 101, 10078–10083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, C.S.; Syljuasen, R.G.; Falck, J.; Schroeder, T.; Ronnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell 2001, 12, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Weinert, T. A DNA damage checkpoint meets the cell cycle engine. Science 1997, 277, 1450–1451. [Google Scholar] [CrossRef]
- Bruinsma, W.; Aprelia, M.; Garcia-Santisteban, I.; Kool, J.; Xu, Y.J.; Medema, R.H. Inhibition of Polo-like kinase 1 during the DNA damage response is mediated through loss of Aurora A recruitment by Bora. Oncogene 2017, 36, 1840–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.H.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E.; et al. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell 1995, 6, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Barlow, J.H.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R., III; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Smith, D.I. Very large common fragile site genes and their potential role in cancer development. Cell. Mol. Life Sci. 2014, 71, 4601–4615. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Wollmuth, C.E.; Das, K.; Hile, S.E.; Regan, S.B.; Barnes, R.P.; Haouzi, A.; Lee, S.M.; House, N.C.M.; Guyumdzhyan, M.; et al. Sequence and Nuclease Requirements for Breakage and Healing of a Structure-Forming (AT)n Sequence within Fragile Site FRA16D. Cell Rep. 2019, 27, 1151–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachages, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Voutsinos, V.; Munk, S.H.N.; Oestergaard, V.H. Common Chromosomal Fragile Sites-Conserved Failure Stories. Genes 2018, 9, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M.; Lowndes, N.F. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Arlt, M.F.; Xu, B.; Durkin, S.G.; Casper, A.M.; Kastan, M.B.; Glover, T.W. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol. Cell. Biol. 2004, 24, 6701–6709. [Google Scholar] [CrossRef] [Green Version]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Durkin, S.G.; Arlt, M.F.; Howlett, N.G.; Glover, T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006, 25, 4381–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and mitosis: A replication legacy. Cell Cycle 2009, 8, 2907–2911. [Google Scholar] [CrossRef]
- Constantinou, A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 2012, 121, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Brosh, R.M., Jr. Holding All the Cards-How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability. Genes 2019, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Schoder, C.; Liehr, T.; Velleuer, E.; Wilhelm, K.; Blaurock, N.; Weise, A.; Mrasek, K. New aspects on chromosomal instability: Chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. Int. J. Oncol. 2010, 36, 307–312. [Google Scholar]
- Schwab, R.A.; Blackford, A.N.; Niedzwiedz, W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010, 29, 806–818. [Google Scholar] [CrossRef]
- Tomida, J.; Itaya, A.; Shigechi, T.; Unno, J.; Uchida, E.; Ikura, M.; Masuda, Y.; Matsuda, S.; Adachi, J.; Kobayashi, M.; et al. A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic Acids Res. 2013, 41, 6930–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pladevall-Morera, D.; Munk, S.; Ingham, A.; Garribba, L.; Albers, E.; Liu, Y.; Olsen, J.V.; Lopez-Contreras, A.J. Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res. 2019, 47, 8004–8018. [Google Scholar] [CrossRef] [Green Version]
- Mohebi, S.; Mizuno, K.; Watson, A.; Carr, A.M.; Murray, J.M. Checkpoints are blind to replication restart and recombination intermediates that result in gross chromosomal rearrangements. Nat. Commun. 2015, 6, 6357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 2005, 121, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.M.; Carr, A.M. Smc5/6: A link between DNA repair and unidirectional replication? Nat. Rev. Mol. Cell Biol. 2008, 9, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, E.C. Suffering in silence: The tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005, 6, 943–953. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Gerlach, V.L. Novel DNA polymerases offer clues to the molecular basis of mutagenesis. Cell 1999, 98, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Despras, E.; Sittewelle, M.; Pouvelle, C.; Delrieu, N.; Cordonnier, A.M.; Kannouche, P.L. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016, 7, 13326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, R.P.; Hile, S.E.; Lee, M.Y.; Eckert, K.A. DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair 2017, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.P.; Tsao, W.C.; Moldovan, G.L.; Eckert, K.A. DNA Polymerase Eta Prevents Tumor Cell-Cycle Arrest and Cell Death during Recovery from Replication Stress. Cancer Res. 2018, 78, 6549–6560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendel, A.; Krijger, P.H.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reissner, T.; Lee, K.Y.; Geacintov, N.E.; Carell, T.; et al. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [Green Version]
- Villa-Hernandez, S.; Bueno, A.; Bermejo, R. The Multiple Roles of Ubiquitylation in Regulating Challenged DNA Replication. Adv. Exp. Med. Biol. 2017, 1042, 395–419. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.; Frattini, C.; Sacristan, M.P.; Gallego-Sanchez, A.; Bermejo, R.; Bueno, A. PCNA Deubiquitylases Control DNA Damage Bypass at Replication Forks. Cell Rep. 2019, 29, 1323–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigaku, Y.; Davies, A.A.; Ulrich, H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010, 465, 951–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rodriguez, N.; Morawska, M.; Wong, R.P.; Daigaku, Y.; Ulrich, H.D. Spatial separation between replisome- and template-induced replication stress signaling. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.P.; Garcia-Rodriguez, N.; Zilio, N.; Hanulova, M.; Ulrich, H.D. Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 2019. [Google Scholar] [CrossRef]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J., Jr.; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 2009, 29, 3344–3354. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.; Andersen, P.L.; Qin, Z.; Xiao, W. Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res. 2013, 41, 2328–2339. [Google Scholar] [CrossRef] [Green Version]
- Boyer, A.S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.S. The human specialized DNA polymerases and non-B DNA: Vital relationships to preserve genome integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef]
- Gallina, I.; Christiansen, S.K.; Pedersen, R.T.; Lisby, M.; Oestergaard, V.H. TopBP1-mediated DNA processing during mitosis. Cell Cycle 2016, 15, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Hellman, A.; Rahat, A.; Scherer, S.W.; Darvasi, A.; Tsui, L.C.; Kerem, B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol. 2000, 20, 4420–4427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, J.; Takahashi, Y.; Goto, J.; Tomiyama, H.; Ishikawa, S.; Yoshino, H.; Minami, N.; Smith, D.I.; Lesage, S.; Aburatani, H.; et al. Mechanisms of genomic instabilities underlying two common fragile-site-associated loci, PARK2 and DMD, in germ cell and cancer cell lines. Am. J. Hum. Genet. 2010, 87, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Schwarzbraun, T.; Speicher, M.R.; Nigg, E.A. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma 2008, 117, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [Green Version]
- West, S.C.; Blanco, M.G.; Chan, Y.W.; Matos, J.; Sarbajna, S.; Wyatt, H.D. Resolution of Recombination Intermediates: Mechanisms and Regulation. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Li, Y.; Truong, L.N.; Shi, L.Z.; Hwang, P.Y.; He, J.; Do, J.; Cho, M.J.; Li, H.; Negrete, A.; et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 2014, 54, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef]
- Guervilly, J.H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Minocherhomji, S.; Hickson, I.D. Structure-specific endonucleases: Guardians of fragile site stability. Trends Cell Biol. 2014, 24, 321–327. [Google Scholar] [CrossRef]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Re, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Garribba, L.; Wu, W.; Ozer, O.; Bhowmick, R.; Hickson, I.D.; Liu, Y. Inducing and Detecting Mitotic DNA Synthesis at Difficult-to-Replicate Loci. Methods Enzymol. 2018, 601, 45–58. [Google Scholar] [CrossRef]
- Falquet, B.; Rass, U. Structure-Specific Endonucleases and the Resolution of Chromosome Underreplication. Genes 2019, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Gallo-Fernandez, M.; Saugar, I.; Ortiz-Bazan, M.A.; Vazquez, M.V.; Tercero, J.A. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 2012, 40, 8325–8335. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Kelly, R.; Martin, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.H.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef]
- Bursomanno, S.; Beli, P.; Khan, A.M.; Minocherhomji, S.; Wagner, S.A.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Hickson, I.D.; Liu, Y. Proteome-wide analysis of SUMO2 targets in response to pathological DNA replication stress in human cells. DNA Repair 2015, 25, 84–96. [Google Scholar] [CrossRef]
- Pardo, B.; Moriel-Carretero, M.; Vicat, T.; Aguilera, A.; Pasero, P. A new role of the Mus81 nuclease for replication completion after fork restart. bioRxiv 2019. [Google Scholar] [CrossRef]
- Ho, C.K.; Mazon, G.; Lam, A.F.; Symington, L.S. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol. Cell 2010, 40, 988–1000. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; McDonald, N.; West, S.C. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 2008, 77, 259–287. [Google Scholar] [CrossRef]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife 2019, 8. [Google Scholar] [CrossRef]
- Priego Moreno, S.; Jones, R.M.; Poovathumkadavil, D.; Scaramuzza, S.; Gambus, A. Mitotic replisome disassembly depends on TRAIP ubiquitin ligase activity. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Biebricher, A.; Hirano, S.; Enzlin, J.H.; Wiechens, N.; Streicher, W.W.; Huttner, D.; Wang, L.H.; Nigg, E.A.; Owen-Hughes, T.; Liu, Y.; et al. PICH: A DNA translocase specially adapted for processing anaphase bridge DNA. Mol. Cell 2013, 51, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef] [Green Version]
- Hengeveld, R.C.; de Boer, H.R.; Schoonen, P.M.; de Vries, E.G.; Lens, S.M.; van Vugt, M.A. Rif1 Is Required for Resolution of Ultrafine DNA Bridges in Anaphase to Ensure Genomic Stability. Dev. Cell 2015, 34, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Hiraga, S.I.; Ly, T.; Garzon, J.; Horejsi, Z.; Ohkubo, Y.N.; Endo, A.; Obuse, C.; Boulton, S.J.; Lamond, A.I.; Donaldson, A.D. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 2017, 18, 403–419. [Google Scholar] [CrossRef]
- Wei, P.C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vidal, A.; Vignard, J.; Mirey, G. Around and beyond 53BP1 Nuclear Bodies. Int. J. Mol. Sci. 2017, 18, 2611. [Google Scholar] [CrossRef] [Green Version]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef]
- Arora, M.; Moser, J.; Phadke, H.; Basha, A.A.; Spencer, S.L. Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep. 2017, 19, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alternative Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [Green Version]
- Ozer, O.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846. [Google Scholar] [CrossRef]
- Medema, R.H.; Lindqvist, A. Boosting and suppressing mitotic phosphorylation. Trends Biochem. Sci. 2011, 36, 578–584. [Google Scholar] [CrossRef]
- Cheng, L.; Hunke, L.; Hardy, C.F. Cell cycle regulation of the Saccharomyces cerevisiae polo-like kinase cdc5p. Mol. Cell. Biol. 1998, 18, 7360–7370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, Y.; Bachant, J.; Wang, H.; Hu, F.; Liu, D.; Tetzlaff, M.; Elledge, S.J. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999, 286, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Ji, J.H.; Choi, Y.C.; Ryu, C.J.; Ko, S.Y. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J. Biol. Chem. 2007, 282, 2473–2482. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Hwang, H.I.; Jang, Y.J. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle 2010, 9, 2389–2398. [Google Scholar] [CrossRef]
- Van Vugt, M.A.; Smits, V.A.; Klompmaker, R.; Medema, R.H. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J. Biol. Chem. 2001, 276, 41656–41660. [Google Scholar] [CrossRef] [Green Version]
- Van Vugt, M.A.; Bras, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004, 117, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Trovesi, C.; Manfrini, N.; Falcettoni, M.; Longhese, M.P. Regulation of the DNA damage response by cyclin-dependent kinases. J. Mol. Biol. 2013, 425, 4756–4766. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Perez, T.; Hayward, D.; Holder, J.; Gruneberg, U.; Barr, F.A. MAD1-dependent recruitment of CDK1-CCNB1 to kinetochores promotes spindle checkpoint signaling. J. Cell Biol. 2019, 218, 1108–1117. [Google Scholar] [CrossRef]
- Bentley, A.M.; Normand, G.; Hoyt, J.; King, R.W. Distinct sequence elements of cyclin B1 promote localization to chromatin, centrosomes, and kinetochores during mitosis. Mol Biol Cell 2007, 18, 4847–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zhang, X.; Jiang, Q.; Clarke, P.R.; Zhang, C. Cyclin B1 is localized to unattached kinetochores and contributes to efficient microtubule attachment and proper chromosome alignment during mitosis. Cell Res. 2008, 18, 268–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, S.Y.; Rosen, E.M.; Jang, Y.J. Novel DNA damage checkpoint in mitosis: Mitotic DNA damage induces re-replication without cell division in various cancer cells. Biochem. Biophys. Res. Commun. 2012, 423, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.A.; Okayasu, R.; Bedford, J.S. Comparison of the induction and disappearance of DNA double strand breaks and γ-H2AX foci after irradiation of chromosomes in G1-phase or in condensed metaphase cells. Mutat. Res. 2008, 639, 108–112. [Google Scholar] [CrossRef]
- Leimbacher, P.A.; Jones, S.E.; Shorrocks, A.K.; de Marco Zompit, M.; Day, M.; Blaauwendraad, J.; Bundschuh, D.; Bonham, S.; Fischer, R.; Fink, D.; et al. MDC1 Interacts with TOPBP1 to Maintain Chromosomal Stability during Mitosis. Mol. Cell 2019, 74, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Giunta, S.; Belotserkovskaya, R.; Jackson, S.P. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010, 190, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Burke, D.J. DNA damage activates the SAC in an ATM/ATR-dependent manner, independently of the kinetochore. PLoS Genet. 2008, 4, e1000015. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, A.; Cole, R.W.; Rieder, C.L. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 2002, 12, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Cesare, A.J.; Hayashi, M.T.; Crabbe, L.; Karlseder, J. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol. Cell 2013, 51, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Hain, K.O.; Colin, D.J.; Rastogi, S.; Allan, L.A.; Clarke, P.R. Prolonged mitotic arrest induces a caspase-dependent DNA damage response at telomeres that determines cell survival. Sci. Rep. 2016, 6, 26766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.T.; Cesare, A.J.; Fitzpatrick, J.A.; Lazzerini-Denchi, E.; Karlseder, J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 2012, 19, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Masamsetti, V.P.; Low, R.R.J.; Mak, K.S.; O’Connor, A.; Riffkin, C.D.; Lamm, N.; Crabbe, L.; Karlseder, J.; Huang, D.C.S.; Hayashi, M.T.; et al. Replication stress induces mitotic death through parallel pathways regulated by WAPL and telomere deprotection. Nat. Commun. 2019, 10, 4224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Z.; Yu, L.; Lin, Y.F.; Matsunaga, S.; Shen, C.Y.; Chen, B.P. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis 2014, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Kabeche, L.; Murnane, J.P.; Zaki, B.I.; Compton, D.A. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014, 4, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, L.; Xu, X.; Li, J.; Stern, D.F. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J. Biol. Chem. 2003, 278, 8468–8475. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.H.; Chou, Y.F.; Ou, Y.H.; Yeh, Y.H.; Tyan, S.W.; Sun, T.P.; Shen, C.Y.; Shieh, S.Y. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. J. Biol. Chem. 2005, 280, 7748–7757. [Google Scholar] [CrossRef] [Green Version]
- Eliezer, Y.; Argaman, L.; Kornowski, M.; Roniger, M.; Goldberg, M. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J. Biol. Chem. 2014, 289, 8182–8193. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Hao, J.; Kong, D.; Cui, X.; Zhang, W.; Wang, H.; Guo, X.; Ma, S.; Liu, X.; Pu, P.; et al. ATM-mediated Mad1 Serine 214 phosphorylation regulates Mad1 dimerization and the spindle assembly checkpoint. Carcinogenesis 2014, 35, 2007–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.; Gatenby, R.; Sidi, S. How Cells Handle DNA Breaks during Mitosis: Detection, Signaling, Repair, and Fate Choice. Cells 2019, 8, 1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Godinez, V.; Wu, T.; Sherman, A.J.; Lee, C.S.; Liaw, L.H.; Zhongsheng, Y.; Yokomori, K.; Berns, M.W. Analysis of DNA double-strand break response and chromatin structure in mitosis using laser microirradiation. Nucleic Acids Res. 2010, 38, e202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.; Hickson, I.D. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin. Cell Dev. Biol. 2011, 22, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Barefield, C.; Karlseder, J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012, 40, 7358–7367. [Google Scholar] [CrossRef]
- Baumann, C.; Korner, R.; Hofmann, K.; Nigg, E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 2007, 128, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, C.F.; Hickson, I.D. PICH promotes mitotic chromosome segregation: Identification of a novel role in rDNA disjunction. Cell Cycle 2016, 15, 2704–2711. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]


{kind=link}
| Cell Cycle Processes Related to Genomic Stability Maintenance and Their CDK-Dependent Regulation | ||
|---|---|---|
| Process | Substrate(s) | Outcome of CDK Phosphorylation |
| Replication origin licensing | MCMs Cdc6 Cdt1 ORC | Impairs loading on chromatin and DNA synthesis initiation in G1 [35] Regulates stabilization and subcellular localization during G1 [36,37] Induces degradation at G1/S [38,39] Inhibits helicase loading [3,40,41] |
| Replication initiation, G1/S-phase transition | MCMs GINS TRESLIN Rb Cdc7 | Activation of MCMs-Cdc45 helicase activity in the S-phase [42,43] Recruitment to chromatin [44] Promotion of DNA replication and association to TopBP1 [45,46,47] Rb inactivation and induction of the S-phase genes transcription in late G1 [48,49,50] Kinase inactivation [51] |
| DNA repair and G2 DNA damage checkpoint recovery (continues on the next page) | 53BP1 RNF8 | Stable binding to Plk1 to silence DNA damage checkpoint and promote entry into mitosis [52,53]; abolition of 53BP1 binding to DSB-flanking chromatin in mitosis [54,55] Impairs RNF8-MDC1 association in mitosis [55] |
| DNA repair and G2 DNA damage checkpoint recovery | TRAIP ATRIP FANCC/FANCG Rad9 RPA Srs2 helicase FOXO1 EXO1 | Activation to promote CMG unloading at stalled replication forks [56] G2/M checkpoint maintenance [57] FA core complex chromatin localization in G2/M [58,59] Interaction with TopBP1 [60] Regulation of DNA repair pathway [61] DSB repair by synthesis-dependent strand annealing [62] Inhibition of FOXO1 induced-apoptosis in presence of DNA damage [63] Regulation of DNA resection and repair pathway choice [64] |
| Mitotic entry | FOXM1 Plk1 Wee1 Myt1 Cdc25A/B/C | Activation of mitotic transcriptional program [65,66] Activation [67] Degradation [68] Inhibition [69] Activation [70] |
| Spindle Assembly Checkpoint (SAC) | Mps1/Bub1 Cdc20 | SAC activation [71,72] APC/C inhibition [73] |
| Homologous recombination (HR) | BRCA2 RECQL4 CtIP Crb2 | Block BRCA2-RAD51 interactions as cells approach mitosis [74] Enhance interaction MRE11/RECQL4 and RECQL4 recruitment to DSBs and stimulated activity [75] Interaction with BRCA1 and NbsI [76,77] Resolution of HR intermediates [78] |
| Non-Homologous End Joining (NHEJ) | XRCC4 | NHEJ suppression in mitosis [79] |
| MiDAS | RECQ5 | Processing of CFSs by Mus81-EME1 in mitosis [80] |
| Holliday Junction resolution | SLX4/Mus81 Gen1 | Formation of the SLX-MUS complex and activation [81,82] Unknown [83] |
| Sister chromatid junction resolution | TopBP1-SLX4 | Promotes TopBP1-SLX4 interaction [84,85] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovejero, S.; Bueno, A.; Sacristán, M.P. Working on Genomic Stability: From the S-Phase to Mitosis. Genes 2020, 11, 225. https://doi.org/10.3390/genes11020225
Ovejero S, Bueno A, Sacristán MP. Working on Genomic Stability: From the S-Phase to Mitosis. Genes. 2020; 11(2):225. https://doi.org/10.3390/genes11020225
Chicago/Turabian StyleOvejero, Sara, Avelino Bueno, and María P. Sacristán. 2020. "Working on Genomic Stability: From the S-Phase to Mitosis" Genes 11, no. 2: 225. https://doi.org/10.3390/genes11020225
APA StyleOvejero, S., Bueno, A., & Sacristán, M. P. (2020). Working on Genomic Stability: From the S-Phase to Mitosis. Genes, 11(2), 225. https://doi.org/10.3390/genes11020225