Network Properties of Cancer Prognostic Gene Signatures in the Human Protein Interactome

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Prognostic Genes and Other Four Gene Sets
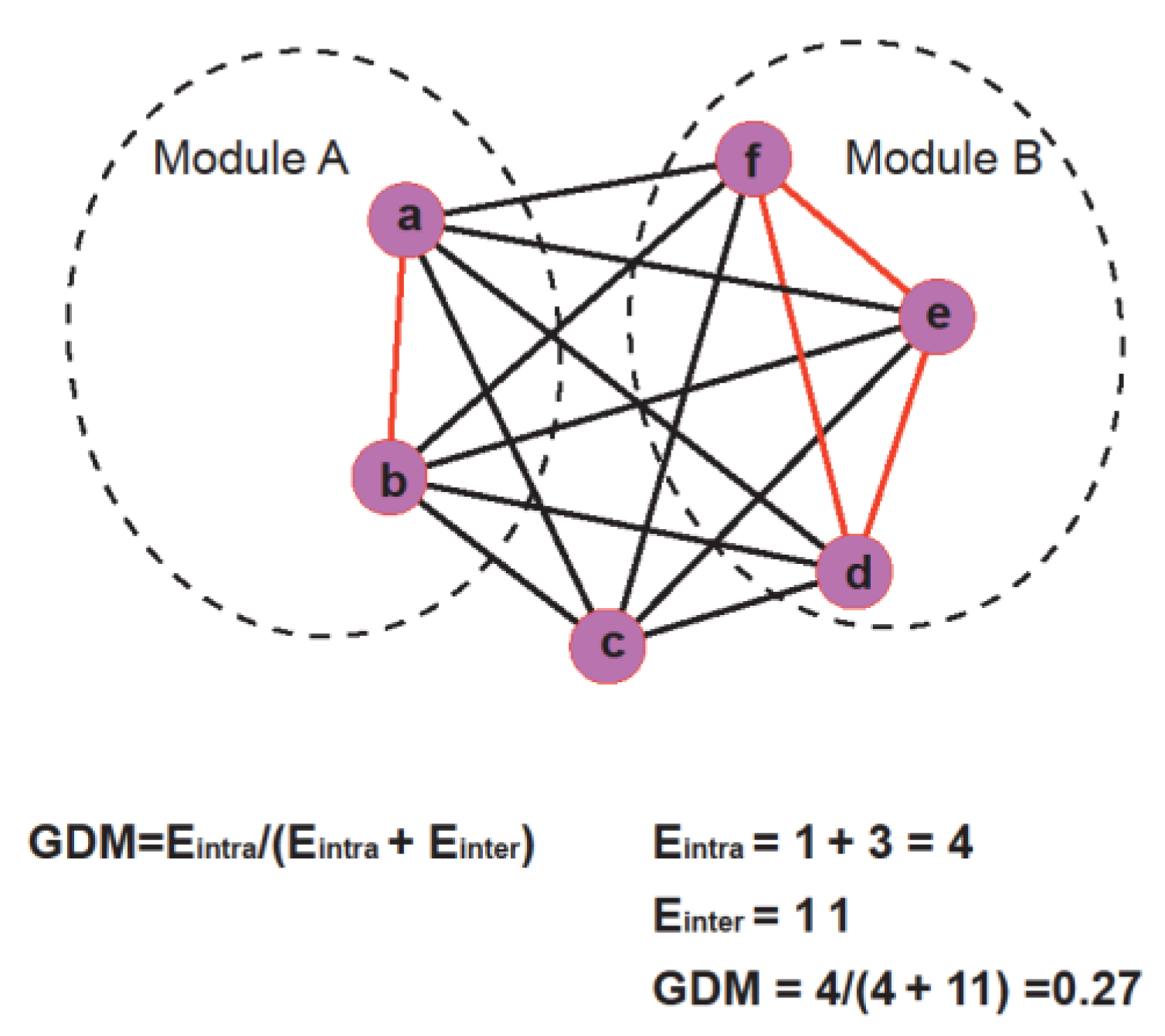
2.2. Biology Networks and Network Modules
2.3. Calculation of Topological Measures
2.4. Functional Enrichment Analysis
3. Results
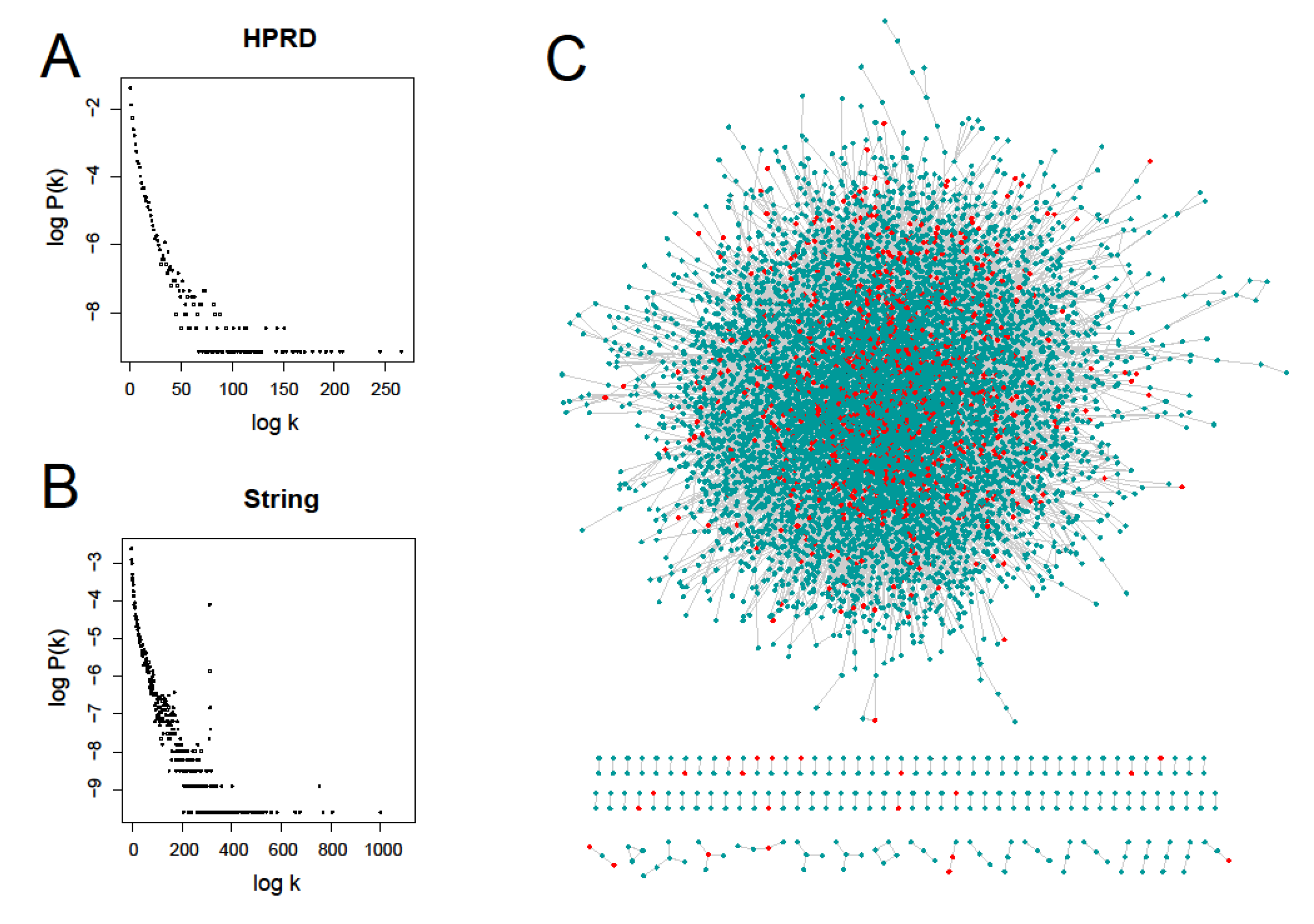
3.1. Overview of Prognostic Genes
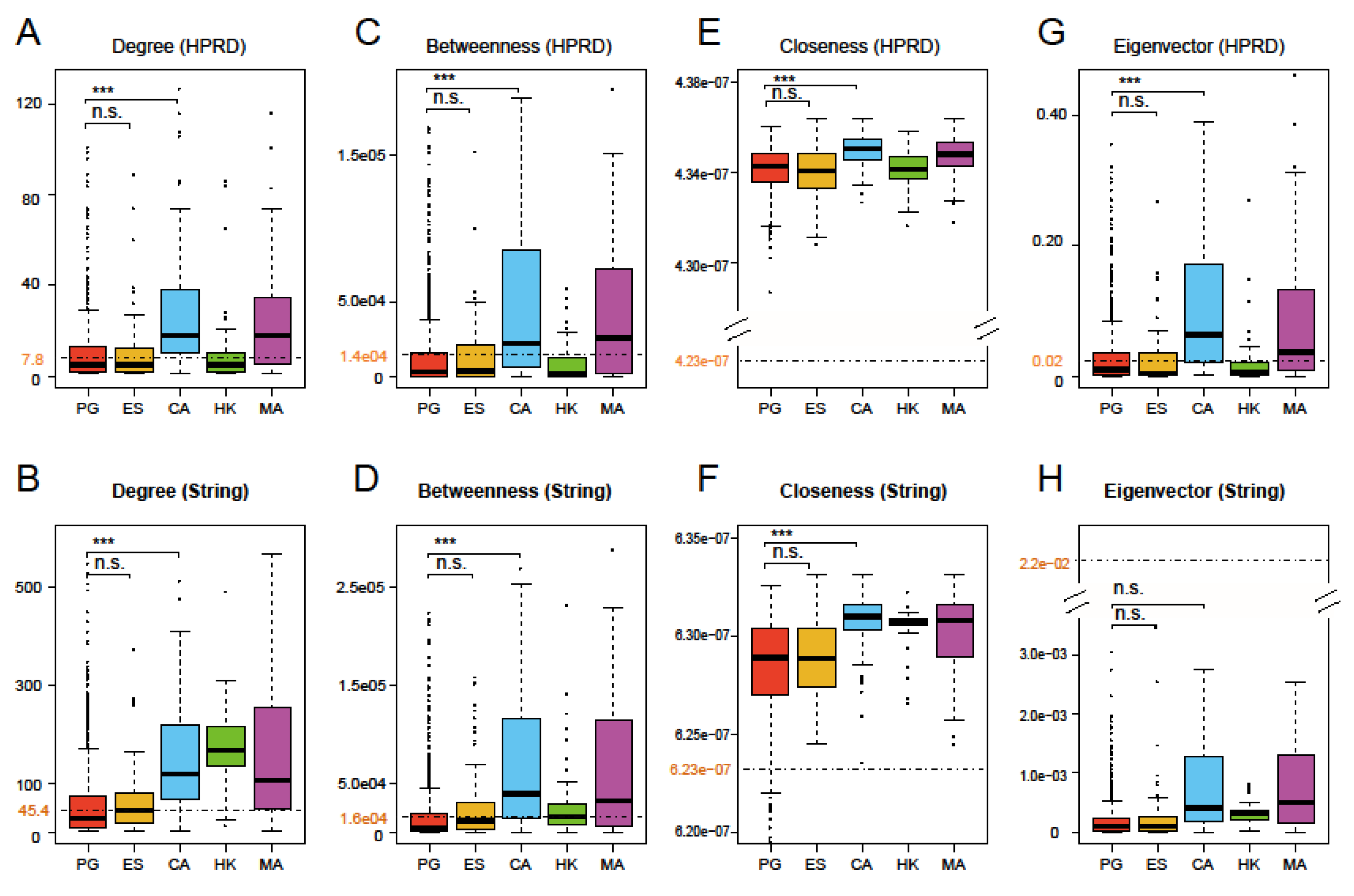
3.2. Four Network Centralities of Prognostic Genes
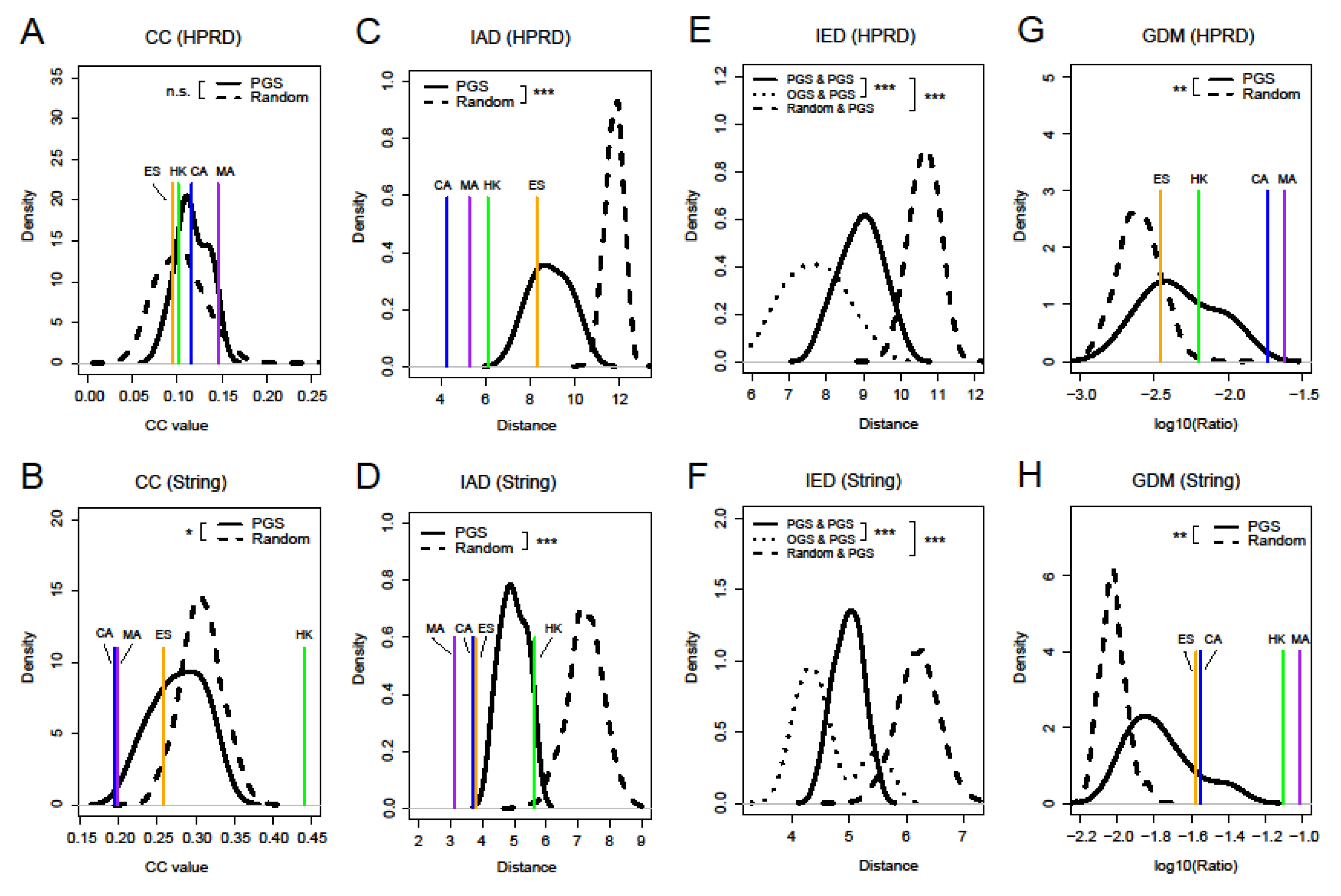
3.3. Four Network Measures of Prognostic Gene Sets
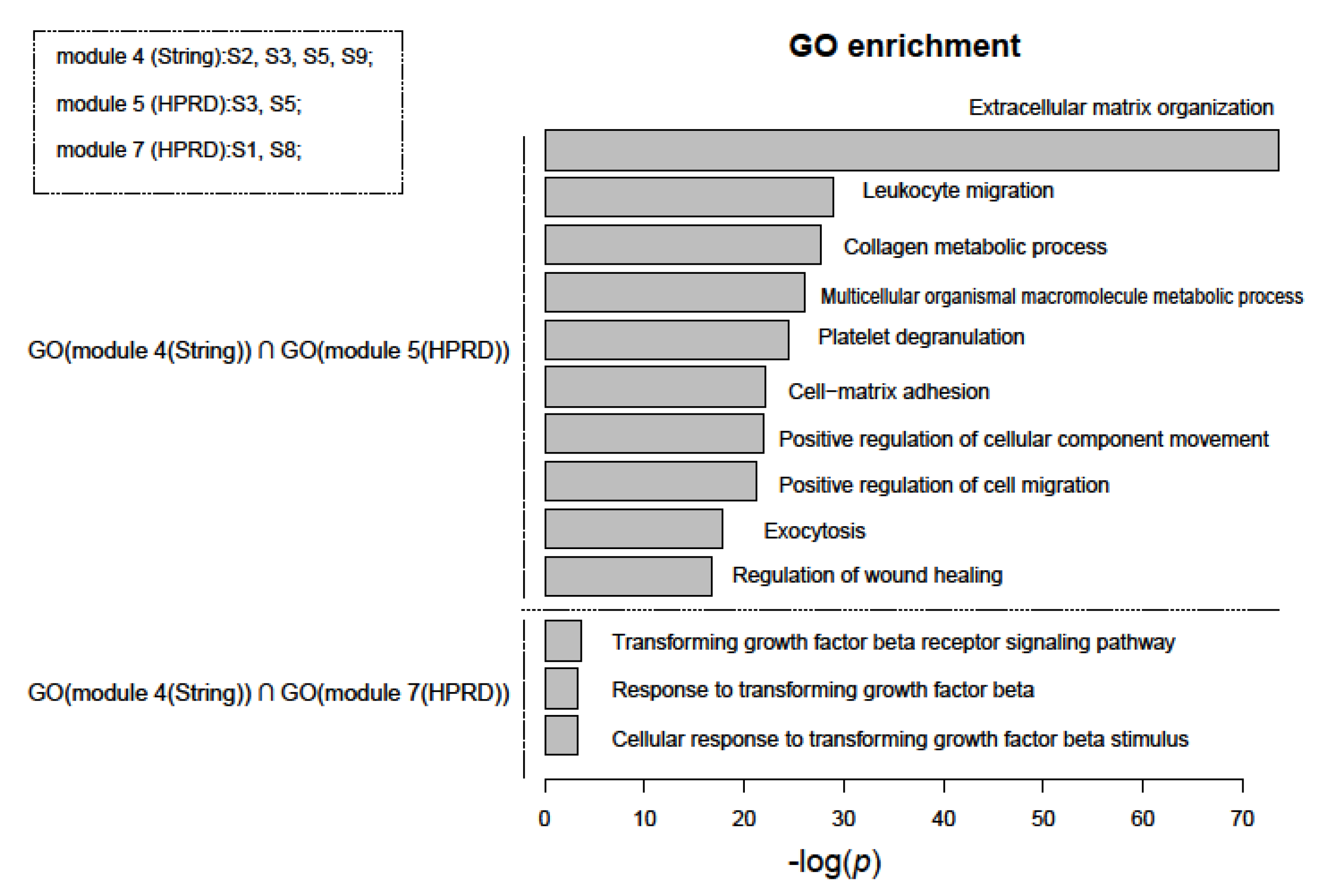
3.4. Functional Analysis of Prognostic Gene Sets
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yuan, Y.; Van Allen, E.M.; Omberg, L.; Wagle, N.; Amin-Mansour, A.; Sokolov, A.; Byers, L.A.; Xu, Y.; Hess, K.R.; Diao, L.; et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat. Biotechnol. 2014, 32, 644–652. [Google Scholar] [CrossRef]
- Li, J.; Lenferink, A.E.; Deng, Y.; Collins, C.; Cui, Q.; Purisima, E.O.; O’Connor-McCourt, M.D.; Wang, E. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat. Commun. 2010, 1, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, E.J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.K.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Bullinger, L.; Dohner, K.; Bair, E.; Frohling, S.; Schlenk, R.F.; Tibshirani, R.; Dohner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Ljungberg, B.; Grankvist, K.; Rasmuson, T.; Tibshirani, R.; Brooks, J.D. Gene expression profiling predicts survival in conventional renal cell carcinoma. PLoS Med. 2006, 3, e13. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Boutros, P.C.; Pintilie, M.; Blackhall, F.H.; Zhu, C.Q.; Strumpf, D.; Johnston, M.R.; Darling, G.; Keshavjee, S.; Waddell, T.K.; et al. Three-gene prognostic classifier for early-stage non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 5562–5569. [Google Scholar] [CrossRef]
- Boutros, P.C.; Lau, S.K.; Pintilie, M.; Liu, N.; Shepherd, F.A.; Der, S.D.; Tsao, M.S.; Penn, L.Z.; Jurisica, I. Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 2824–2828. [Google Scholar] [CrossRef] [Green Version]
- Sveen, A.; Agesen, T.H.; Nesbakken, A.; Meling, G.I.; Rognum, T.O.; Liestol, K.; Skotheim, R.I.; Lothe, R.A. ColoGuidePro: A prognostic 7-gene expression signature for stage III colorectal cancer patients. Clin. Cancer Res. 2012, 18, 6001–6010. [Google Scholar] [CrossRef] [Green Version]
- Gerami, P.; Cook, R.W.; Wilkinson, J.; Russell, M.C.; Dhillon, N.; Amaria, R.N.; Gonzalez, R.; Lyle, S.; Johnson, C.E.; Oelschlager, K.M.; et al. Development of a prognostic genetic signature to predict the metastatic risk associated with cutaneous melanoma. Clin. Cancer Res. 2015, 21, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Weigel, M.T.; Dowsett, M. Current and emerging biomarkers in breast cancer: Prognosis and prediction. Endocr. Relat. Cancer 2010, 17, R245–R262. [Google Scholar] [CrossRef] [PubMed]
- Furlong, L.I. Human diseases through the lens of network biology. Trends Genet. 2013, 29, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Dong, X.; Fu, Y.; Tian, W. An iterative network partition algorithm for accurate identification of dense network modules. Nucleic Acids Res. 2012, 40, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, H.; Howton, T.C.; Sun, Y.; Weinberger, N.; Belkhadir, Y.; Mukhtar, M.S. Network biology discovers pathogen contact points in host protein-protein interactomes. Nat. Commun. 2018, 9, 2312. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, M.D.; Vandin, F.; Wu, H.T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Liu, Q.; Zhou, Y.; Tao, C.; Zhao, Z.; Sun, J.; Xu, H. Oncogenes and tumor suppressor genes: Comparative genomics and network perspectives. BMC Genom. 2015, 16, S8. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhao, Z. A comparative study of cancer proteins in the human protein-protein interaction network. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [Green Version]
- Ein-Dor, L.; Zuk, O.; Domany, E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 5923–5928. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ledesma, E.; Verhaak, R.G.; Trevino, V. Identification of a multi-cancer gene expression biomarker for cancer clinical outcomes using a network-based algorithm. Sci. Rep. 2015, 5, 11966. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HARLOZINSKA, A. Progress in molecular mechanisms of tumor metastasis and angiogenesis. Anticancer Res. 2005, 25, 3327–3333. [Google Scholar]
- Craft, P.; Harris, A. Clinical prognostic significance of tumour angiogenesis. Ann. Oncol. 1994, 5, 305–311. [Google Scholar] [CrossRef]
- Schliephake, H. Prognostic relevance of molecular markers of oral cancer—A review. Int. J. Oral Maxillofac. Surg. 2003, 32, 233–245. [Google Scholar] [CrossRef]
- Gray, K.A.; Daugherty, L.C.; Gordon, S.M.; Seal, R.L.; Wright, M.W.; Bruford, E.A. Genenames.org: The HGNC resources in 2013. Nucleic Acids Res. 2013, 41, D545–D552. [Google Scholar] [CrossRef] [Green Version]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, P.; Caflisch, A. Multistep greedy algorithm identifies community structure in real-world and computer-generated networks. Phys. Rev. E 2008, 78, 026112. [Google Scholar] [CrossRef] [Green Version]
- Ozgur, A.; Vu, T.; Erkan, G.; Radev, D.R. Identifying gene-disease associations using centrality on a literature mined gene-interaction network. Bioinformatics 2008, 24, i277–i285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Mondragon, R.J. Accurately modeling the internet topology. Phys. Rev. E 2004, 70, 066108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, P.F.; Bates, P.A. Global topological features of cancer proteins in the human interactome. Bioinformatics 2006, 22, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Barter, R.L.; Schramm, S.-J.; Mann, G.J.; Yang, Y.H. Network-based biomarkers enhance classical approaches to prognostic gene expression signatures. BMC Syst. Biol. 2014, 8, S5. [Google Scholar] [CrossRef] [Green Version]
- Boccaletti, S.; Latora, V.; Moreno, Y.; Chavez, M.; Hwang, D.-U. Complex networks: Structure and dynamics. Phys. Rep. 2006, 424, 175–308. [Google Scholar] [CrossRef]
- De Jonge, H.J.; Fehrmann, R.S.; de Bont, E.S.; Hofstra, R.M.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.; van der Zee, A.G.; te Meerman, G.J.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [Green Version]
- Latora, V.; Marchiori, M. Efficient behavior of small-world networks. Phys. Rev. Lett. 2001, 87, 198701. [Google Scholar] [CrossRef] [Green Version]
- Sharan, R.; Ulitsky, I.; Shamir, R. Network-based prediction of protein function. Mol. Syst. Biol. 2007, 3, 88. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangaletti, S.; Chiodoni, C.; Tripodo, C.; Colombo, M.P. The good and bad of targeting cancer-associated extracellular matrix. Curr. Opin. Pharmacol. 2017, 35, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Blobe, G.C. Role of transforming growth factor Beta in human cancer. J. Clin. Oncol. 2005, 23, 2078–2093. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.C.; Lin, C.C.; Chang, J.Y.; Mori, H.; Juan, H.F.; Huang, H.C. Predicting essential genes based on network and sequence analysis. Mol. Biosyst. 2009, 5, 1672–1678. [Google Scholar] [CrossRef]
- Samal, A.; Singh, S.; Giri, V.; Krishna, S.; Raghuram, N.; Jain, S. Low degree metabolites explain essential reactions and enhance modularity in biological networks. BMC Bioinform. 2006, 7, 118. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.Y.; Zhang, J. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 6987–6992. [Google Scholar] [CrossRef] [Green Version]
- Zetter, B.R. Angiogenesis and tumor metastasis. Annu. Rev. Med. 1998, 49, 407–424. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Zuo, S.; Dai, G.; Ren, X. Identification of a 6-gene signature predicting prognosis for colorectal cancer. Cancer Cell Int. 2019, 19, 6. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, I.W.; Linding, R.; Warde-Farley, D.; Liu, Y.; Pesquita, C.; Faria, D.; Bull, S.; Pawson, T.; Morris, Q.; Wrana, J.L. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009, 27, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Stein, L. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 2012, 13, R112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Yang, Z.; Xie, L.; Han, L.; Qu, X.; Yang, Y.; Zhang, Y.; He, Z.; Wang, Y.; Li, J. Circular RNAs: Regulators of Cancer-Related Signaling Pathways and Potential Diagnostic Biomarkers for Human Cancers. Theranostics 2017, 7, 3106–3117. [Google Scholar] [CrossRef]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study 1 | Disease | Number of Prognostic Genes in Study | Gene Set | Number of Prognostic Genes in Gene Set |
|---|---|---|---|---|
| Gentles et al. (Nat. Med. 2015) | Multiple tumor types | various | S1 | 120 * |
| The Cancer Genome Atlas Research Network (Nature. 2011) | Ovarian carcinoma | 190 | S2 | 185 |
| Lenz et al. (N. Engl. J. Med. 2008) | (Diffuse) Large-B-cell lymphomas | 39,283,71 | S3 | 330 |
| Zhao et al. (PLoS Med. 2006) | Renal cell carcinoma | 259 | S4 | 222 |
| Dave et al. (N. Engl. J. Med. 2006) | Burkitt’s lymphoma | 217 | S5 | 200 |
| Bullinger et al. (N. Engl. J. Med. 2004) | Acute myeloid leukemia (AML) | 133 | S6 | 103 |
| Liu et al. (J. Natl. Cancer Inst. 2014) | (Triple-negative) Breast cancer | 11 | S7 | 135 |
| Wang et al. (Lancet. 2005) | (Lymph-node-negative) Breast cancer | 76 | ||
| van de Vijveret al. (N. Engl. J. Med. 2002) | Breast cancer | 70 | ||
| Wistuba et al. (Clin. Cancer Res. 2013) | Lung adenocarcinoma | 31 | S8 | 118 |
| Tang et al. (Clin. Cancer Res. 2013) | Non-small cell lung cancer (NSCLC) | 12 | ||
| Xie et al. (Clin. Cancer Res. 2011) | NSCLC | 59 | ||
| Zhu et al. (J. Clin. Oncol. 2010) | NSCLC | 15 | ||
| Boutros et al. (Proc. Natl. Acad. Sci. USA 2009) | NSCLC | 6 | ||
| Lau et al. (J. Clin. Oncol. 2007) | NSCLC | 3 | ||
| Gerami et al. (Clin. Cancer Res. 2015) | Melanoma | 28 | S9 | 174 |
| Wu et al. (Proc. Natl. Acad. Sci. USA 2013) | Prostate cancer | 32 | ||
| Li et al. (J. Clin. Oncol. 2013) | AML | 24 | ||
| Lohavanichbutr et al. (Clin. Cancer Res. 2013) | Oral squamous cell carcinomas (OSCC) | 13 | ||
| Sveen et al. (Clin. Cancer Res. 2012) | Colorectal cancer | 7 | ||
| Smith et al. (Gastroenterology. 2010) | Colon cancer | 34 | ||
| Ramaswamy et al. (Nat. Genet. 2003) | Solid tumors | 17 | ||
| Yeoh et al. (Cancer Cell. 2002) | Acute lymphoblastic leukemia (ALL) | 7–20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yan, S.; Jiang, C.; Ji, Z.; Wang, C.; Tian, W. Network Properties of Cancer Prognostic Gene Signatures in the Human Protein Interactome. Genes 2020, 11, 247. https://doi.org/10.3390/genes11030247
Zhang J, Yan S, Jiang C, Ji Z, Wang C, Tian W. Network Properties of Cancer Prognostic Gene Signatures in the Human Protein Interactome. Genes. 2020; 11(3):247. https://doi.org/10.3390/genes11030247
Chicago/Turabian StyleZhang, Jifeng, Shoubao Yan, Cheng Jiang, Zhicheng Ji, Chenrun Wang, and Weidong Tian. 2020. "Network Properties of Cancer Prognostic Gene Signatures in the Human Protein Interactome" Genes 11, no. 3: 247. https://doi.org/10.3390/genes11030247
APA StyleZhang, J., Yan, S., Jiang, C., Ji, Z., Wang, C., & Tian, W. (2020). Network Properties of Cancer Prognostic Gene Signatures in the Human Protein Interactome. Genes, 11(3), 247. https://doi.org/10.3390/genes11030247