Effect of Transgenesis on mRNA and miRNA Profiles in Cucumber Fruits Expressing Thaumatin II

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA and RNA Extraction
2.3. Confirmation of Transgenicity
2.4. Library Construction and Sequencing
2.5. Bioinformatic Analysis
2.6. Validation of DEGs Expression Profiles by qPCR
3. Results
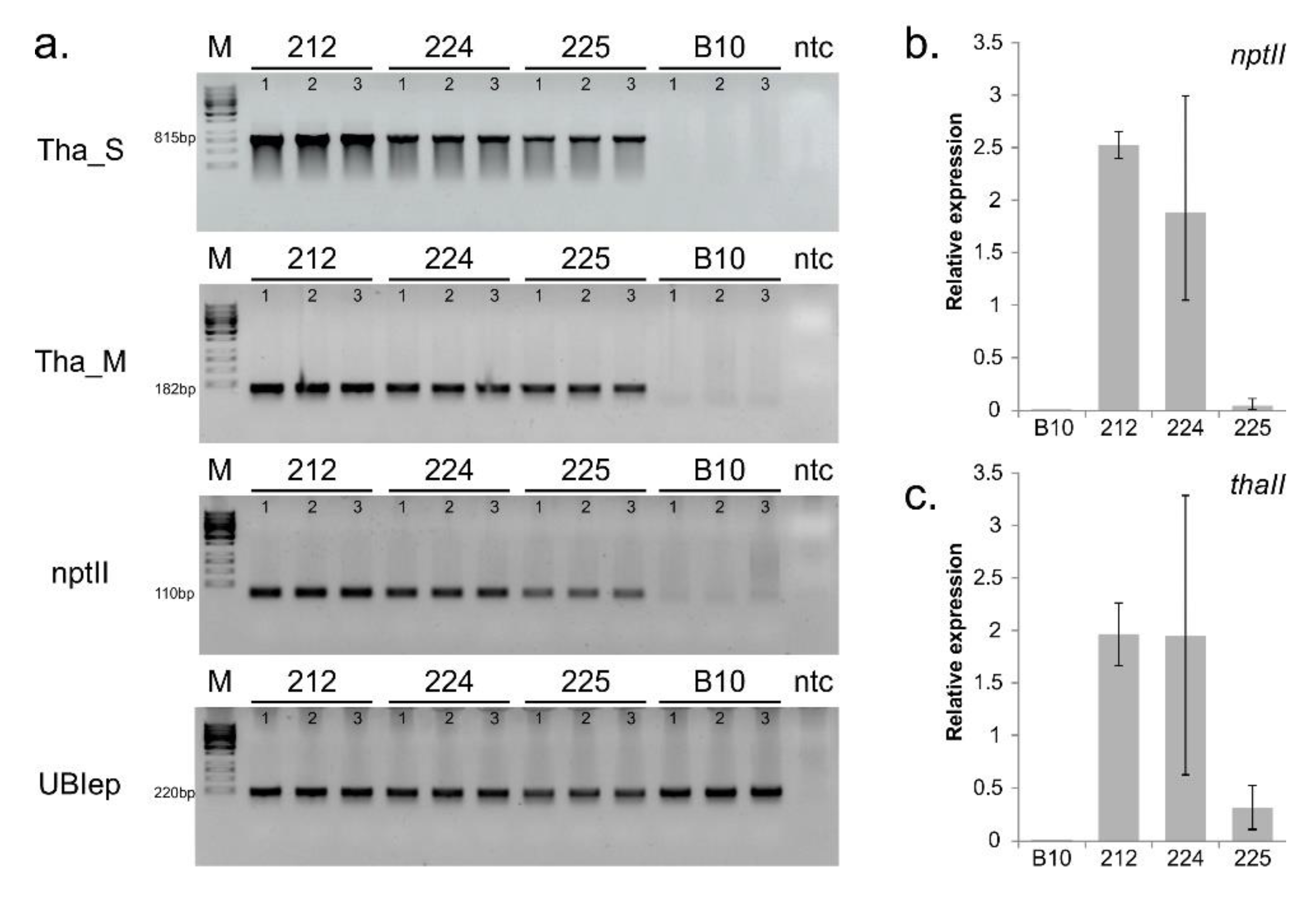
3.1. Confirmation of Transgenes Stability and Expression
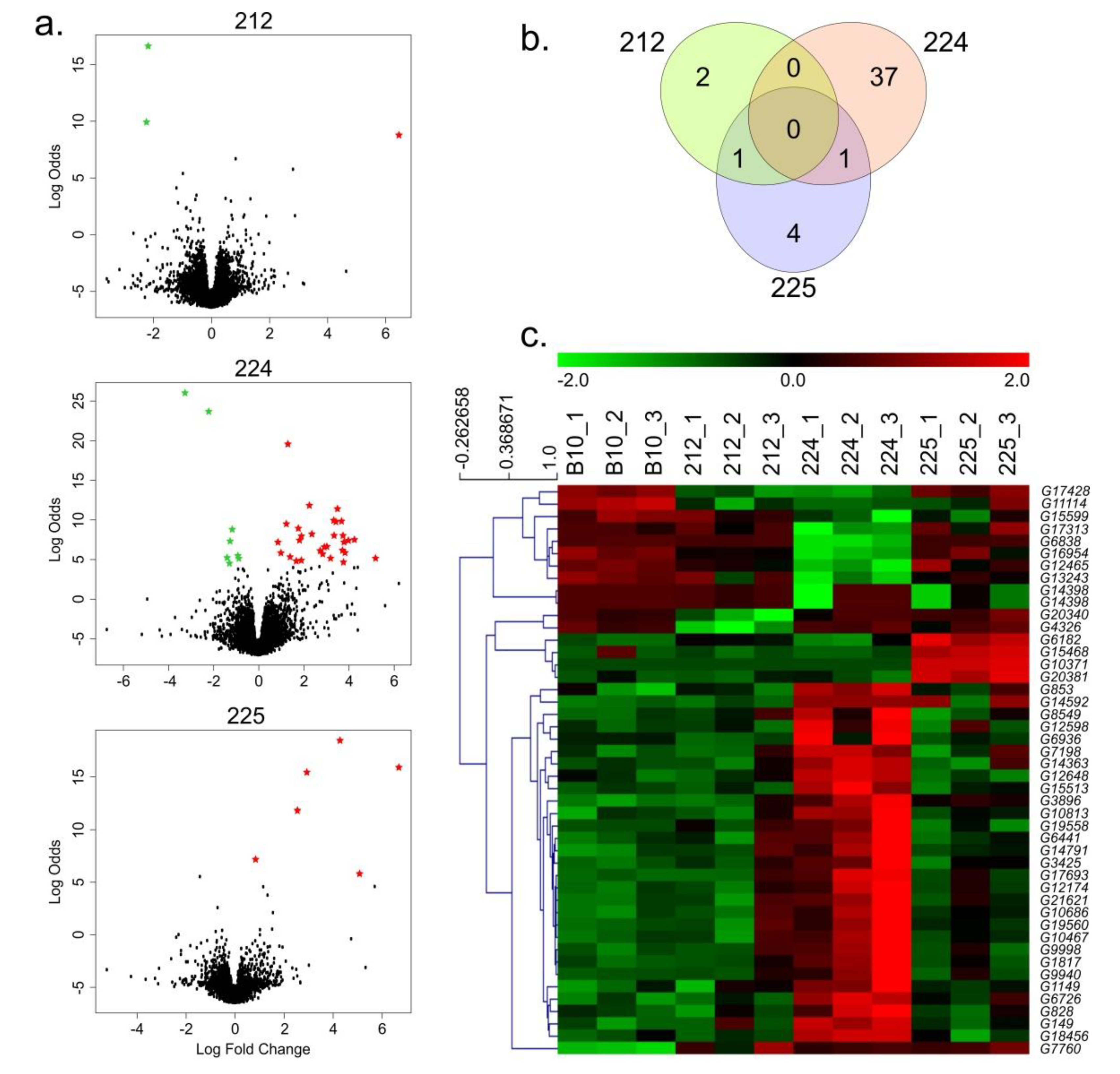
3.2. Identification of DEGs
3.3. DEGs in the Transgenic 212 Line
3.4. DEGs in the Transgenic 224 Line
3.5. DEGs in the Transgenic 225 Line
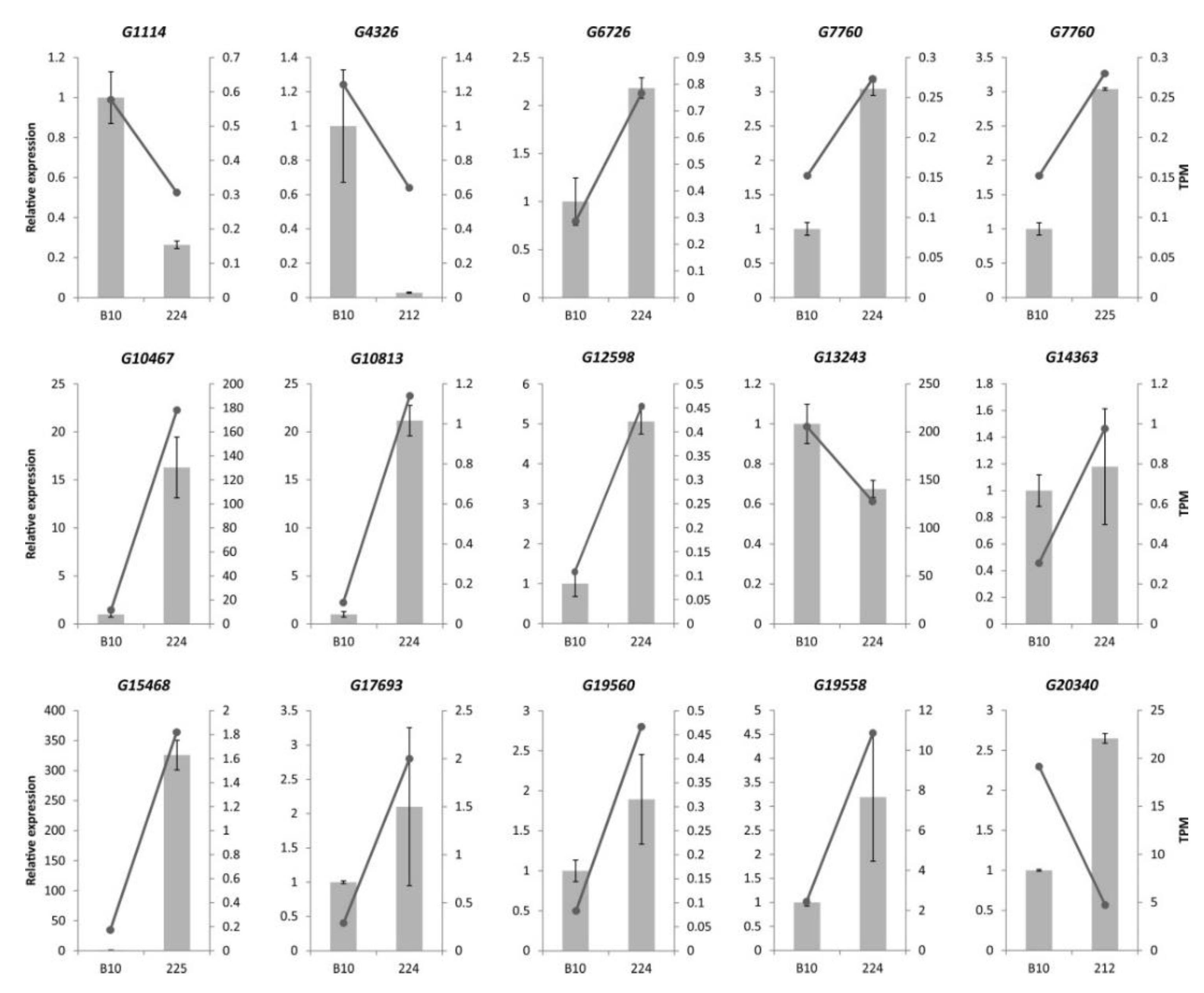
3.6. Confirmation of Illumina RNA-seq Expression by qPCR
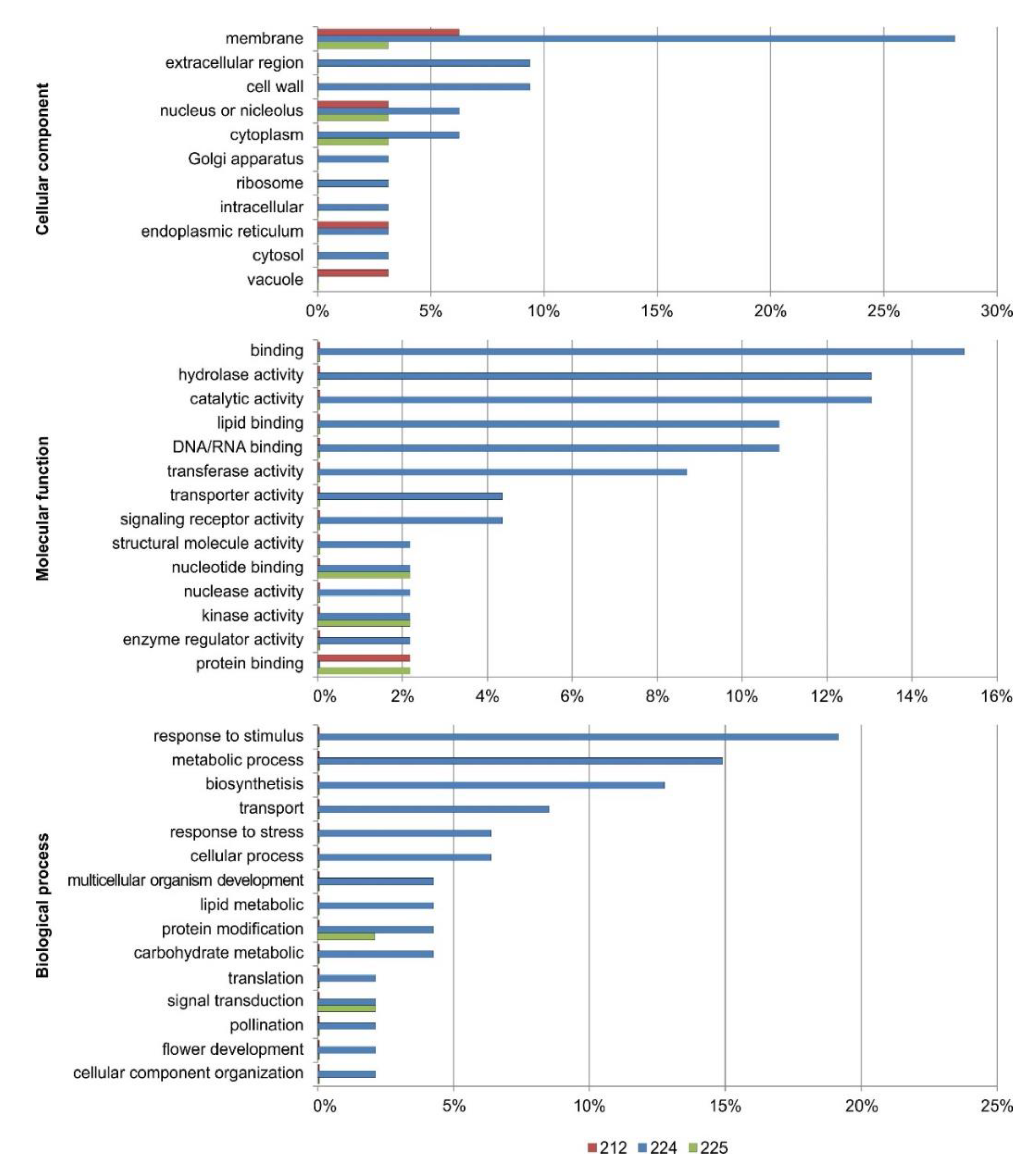
3.7. Functional Annotation of the DEGs
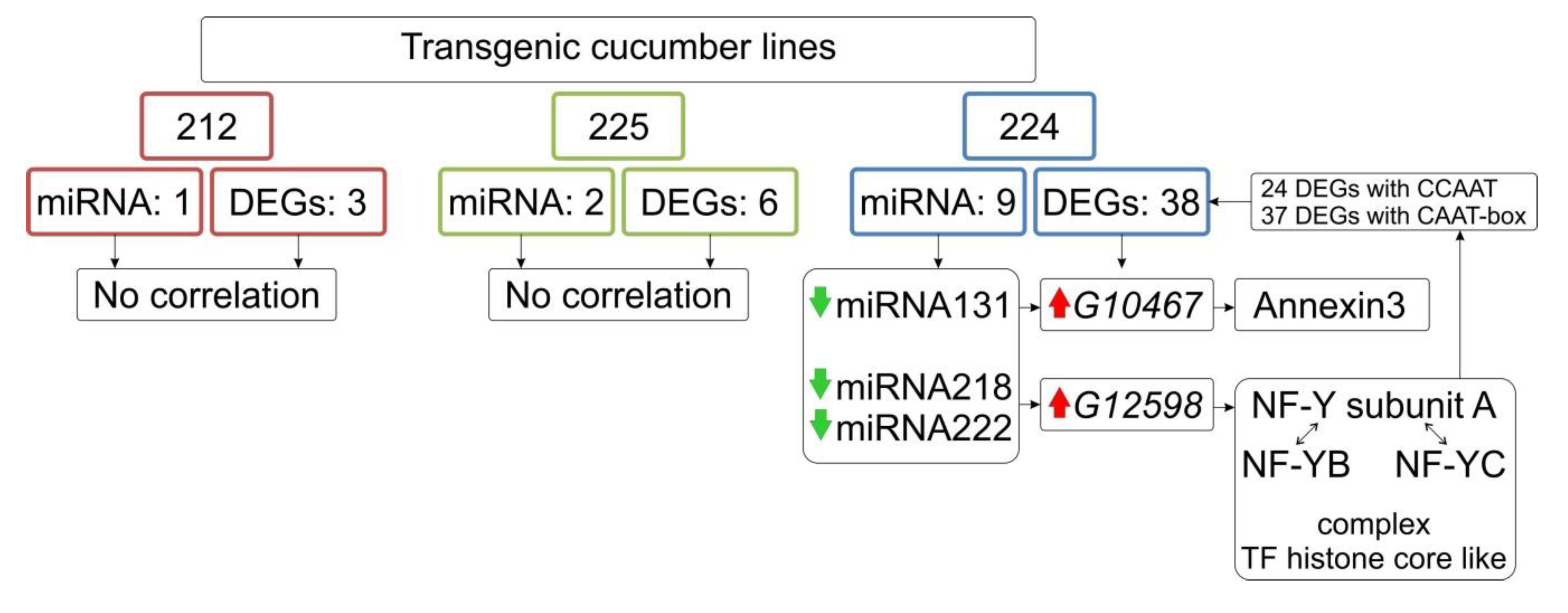
3.8. Identification of Differentially Expressed miRNA
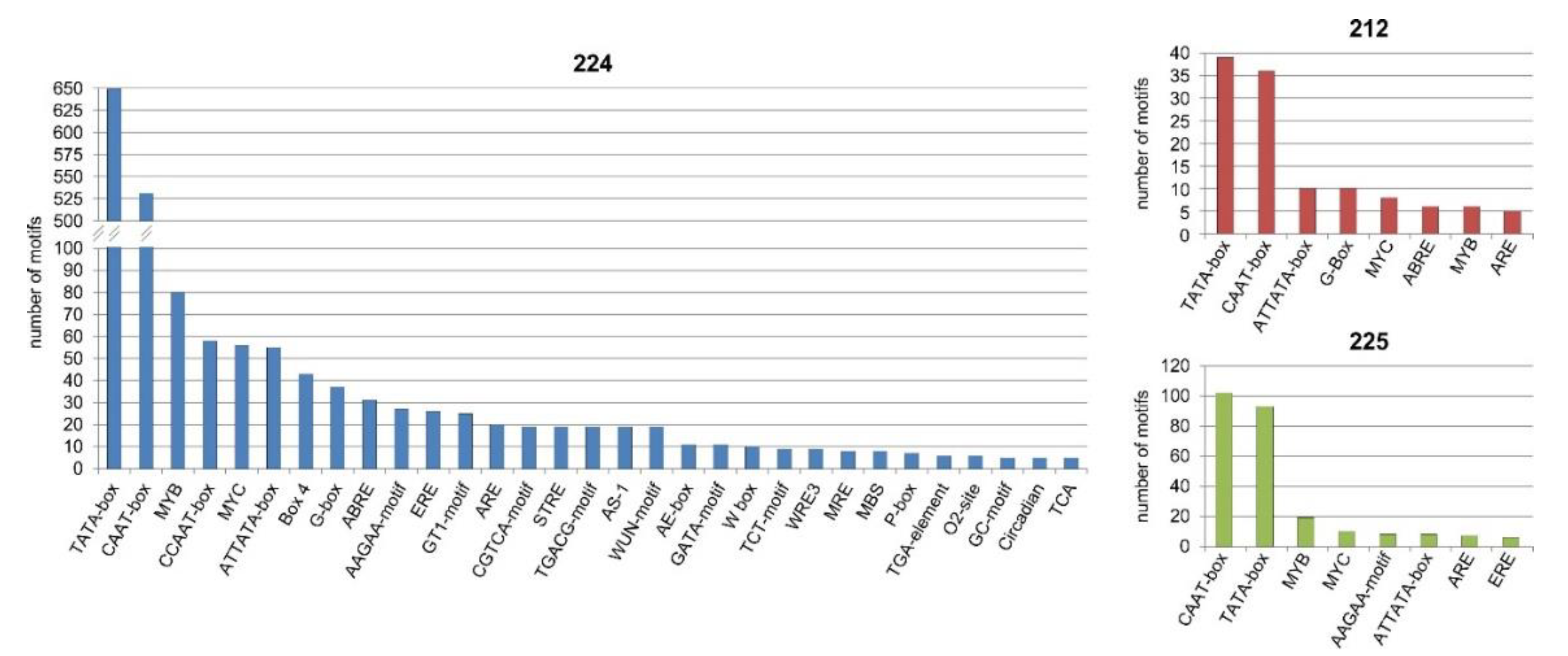
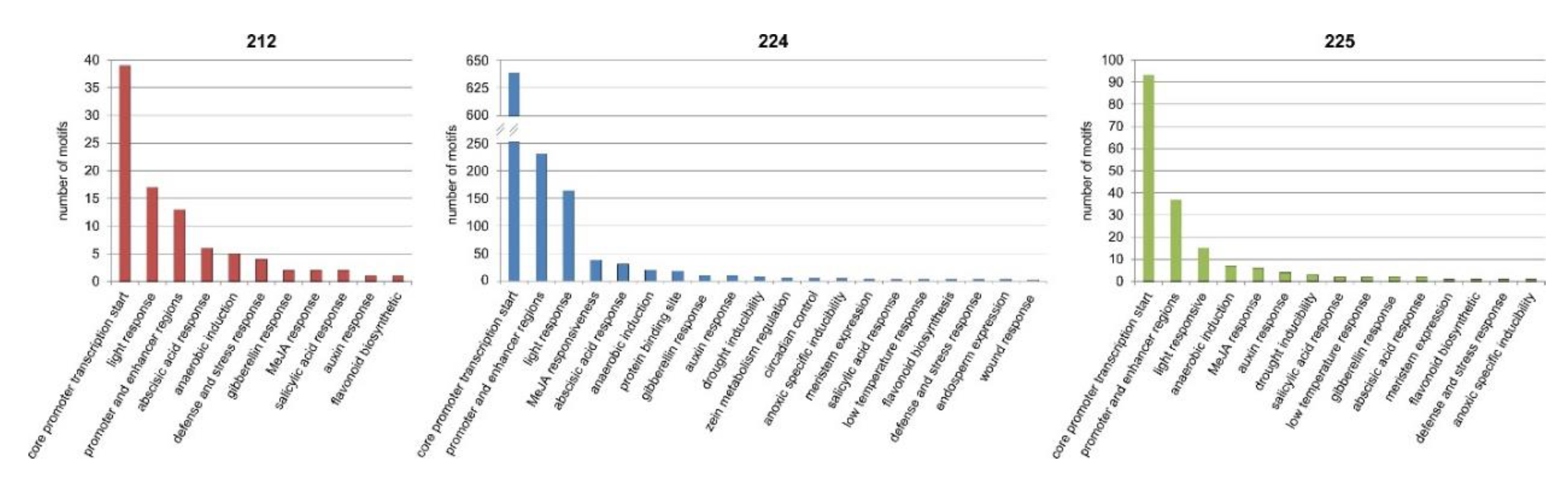
3.9. Bioinformatics Analysis of the Upstream Promoter Regions of the DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szwacka, M.; Morawski, M.; Burza, W. Agrobacterium Tumefaciens-Mediated Cucumber Transformation with Thaumatin II cDNA. J. Appl. Genet. 1996, 37A, 126–129. [Google Scholar]
- Chinedu, S.N.; Oluwadamisi, A.Y.; Popoola, S.T.; Bolaji, J.D.; Epelle, T. Analyses of the Leaf, Fruit and Seed of Thaumatococcus Daniellii (Benth.): Exploring Potential Uses. Pak. J. Bot. 2014, 17, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Van Der Well, H.; Bel, W.J. Enzymatic Properties of the Sweet-Tasting Proteins Thaumatin and Monellin after Partial Reduction. Eur. J. Biochem. 1980, 418, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, R.; Kitabatake, N. Structure-Sweetness Relationship in Thaumatin: Importance of Lysine Residues. Chem. Senses 2001, 26, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, L.; Arró, M.; Hoebeke, J.; Meeks-Wagner, D.R.; Van, K.T.T. Immunological evidence of thaumatin-like proteins during tobacco floral differentiation. Plant Physiol. 1992, 98, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fils-Lycaon, B.R.; Wiersma, P.A.; Eastwell, K.C.; Sautiere, P. A cherry protein and its gene, abundantly expressed in ripening fruit, have been identified as thaumatin-like. Plant Physiol. 1996, 111, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.J.; Sturrock, R.; Ekramoddoullah, A.K. The superfamily of thaumatin-like proteins: Its origin, evolution, and expression towards biological function. Plant Cell Rep. 2010, 29, 419–436. [Google Scholar] [CrossRef]
- Trudel, J.; Grenier, J.; Potvin, C.; Asselin, A. Several Thaumatin-like Proteins Bind to 1,3-Glucans. Plant Physiol. 1998, 118, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.; Kearsley, M.W. Sweeteners and Sugar Alternatives in Food Technology, 2nd ed.; Wiley-Blackwell: London, UK, 2012; pp. 215–218. [Google Scholar] [CrossRef]
- Szwacka, M.; Burza, W.; Zawirska-Wojtasiak, R.; Gośliński, M. Genetically Modified Crops Expressing 35S-Thaumatin II Transgene: Sensory Properties and Food Safety Aspects. Compr. Rev. 2012, 11, 174–186. [Google Scholar] [CrossRef]
- Witty, M. Preprothaumatin II is processed to biological activity in Solanum tuberosum. Biotechnol. Lett. 1990, 12, 131–136. [Google Scholar] [CrossRef]
- Szwacka, M.; Krzymowska, M.; Osuch, A.; Kowalczyk, M.E.; Malepszy, S. Variable properties of transgenic cucumber plants containing the thaumatin II gene from Thaumatococcus daniellii. Acta Physiol. Plant 2002, 24, 173–185. [Google Scholar] [CrossRef]
- Bartoszewski, G.; Niedziela, A.; Szwacka, M.; Niemirowicz-Szczytt, K. Modification of tomato taste in transgenic plants carrying a thaumatin gene from Thaumatococcus daniellii Benth. Plant Breed. 2003, 122, 347–351. [Google Scholar] [CrossRef]
- Korneeva, I.; Firsov, A.; Lebedev, V.; Schestibratov, K.; Pushin, A.; Shulga, O.; Dolgov, S. Expression and subcellular localization of PR-5 protein with different signal sequences in transgenic tomato and tobacco plants. Rev. Cytol. Biol. Veg. Bot. 2005, 28, e267. [Google Scholar]
- Dolgov, J.S.V.; Lebedev, V.G.; Firsov, A.P. Pear fruit taste modification by thaumatin II gene expression. In XI International Pear Symposium; International Society for Horticultural Science, Acta Horticulturae: Leuven, Belgium, 2011; Volume 909, pp. 67–73. [Google Scholar] [CrossRef]
- Sidorova, T.; Miroshnichenko, D.; Tjukavin, G.; Dolgov, S. Expression of thaumatin II gene in carrot for improved pathogen resistance. Acta Hortic. 2012, 929, 357–364. [Google Scholar] [CrossRef]
- Schestibratov, J.K.A.; Dolgov, S.V. Transgenic strawberry plants expressing a thaumatin II gene demonstrate enhanced resistance to Botrytis cinerea. Sci. Hortic. 2005, 106, 177–189. [Google Scholar] [CrossRef]
- Dolgov, J.S.V.; Schestibratov, K.A.; Mikhailov, R.V. Apple Transformation with the gene of supersweet protein Thaumatin II. Acta Hortic. 2004, 663, 507–510. [Google Scholar] [CrossRef]
- Tagashira, N.; Plader, W.; Filipecki, M.; Yin, Z.; Wisniewska, A.; Gaj, P.; Szwacka, M.; Fiehn, O.; Hoshi, Y.; Kondo, K.; et al. The metabolic profiles of transgenic cucumber lines vary with different chromosomal locations of the transgene. Cell. Mol. Biol. Lett. 2005, 10, 697. [Google Scholar] [PubMed]
- Filipecki, M.; Malepszy, S. Unintended consequences of plant transformation: A molecular insight. J. Appl. Genet. 2006, 47, 277–286. [Google Scholar] [CrossRef]
- Zawirska-Wojtasiak, R.; Gośliński, M.; Szwacka, M.; Gajc-Wolska, J.; Mildner-Szkudlarz, S. Aroma evaluation of transgenic, Thaumatin II-producing cucumber fruits. Food Chem. 2009, 74, 204–210. [Google Scholar] [CrossRef]
- Szwacka, M.; Tykarska, T.; Wisniewska, A.; Kuras, M.; Bilski, H.; Malepszy, S. Leaf morphology and anatomy of transgenic cucumber lines tolerant to downy mildew. Biol. Plant. 2009, 53, 697–701. [Google Scholar] [CrossRef]
- Kosieradzka, I.; Sawosz, E.; Pastuszewska, B.; Szwacka, M.; Malepszy, S.; Bielecki, W.; Czuminska, K. The effect of feeding diets with genetically modified cucumbers on the growth and health status of rats. J. Anim. Feed Sci. 2001, 10, 7–12. [Google Scholar] [CrossRef]
- Masuda, T.; Taguchi, W.; Sano, A.; Ohta, K.; Kitabatake, N.; Tani, F. Five amino acid residues in cysteine-rich domain of human T1R3 were involved in the response for sweet-tasting protein, thaumatin. Biochimie 2013, 95, 1502–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 March 2020).
- Osipowski, P.; Pawełkowicz, M.; Wojcieszek, M.; Skarzyńska, A.; Przybecki, Z.; Pląder, W. A high-quality cucumber genome assembly enhances computational comparative genomics. Mol. Genet. Genom. 2020, 295, 177–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Dong, X.; Tian, L.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- MeV. Available online: https://sourceforge.net/projects/mev-tm4/ (accessed on 19 March 2020).
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Pfam. Available online: https://pfam.xfam.org (accessed on 19 March 2020).
- InterPro Classification of Protein Families. Available online: https://www.ebi.ac.uk/interpro (accessed on 19 March 2020).
- PROSITE. Available online: https://prosite.expasy.org (accessed on 19 March 2020).
- TargetP. Available online: http://www.cbs.dtu.dk/services/TargetP/ (accessed on 19 March 2020).
- WoLF PSORT. Available online: https://wolfpsort.hgc.jp/ (accessed on 19 March 2020).
- TmHMM. Available online: https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 (accessed on 19 March 2020).
- miRBase: The microRNA Database. Available online: http://www.mirbase.org/ (accessed on 19 March 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- psRNAtarget. Available online: http://plantgrn.noble.org/psRNATarget/ (accessed on 19 March 2020).
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Skarzyńska, A.; Pawełkowicz, M.; Pląder, W.; Przybecki, Z. The utility of optical detection system (qPCR) and bioinformatics methods in reference gene expression analysis. In: Photonics Applications in Astronomy, Communications, Industry, and High-Energy Physics Experiments 2016. Int. Soc. Opt. Photonics 2016, 1003130. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.L.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Pape, S.L. EasyqpcR: EasyqpcR for easy analysis of real-time PCR data at IRTOMITINSERM U1082. Irtomit-Inser. U 2012, 1082. [Google Scholar] [CrossRef]
- Szwacka, M.; Siedlecka, E.; Malepszy, S. Expression pattern of the pre-prothaumatin ii gene under the control of the CaMV 35s promoter in transgenic cucumber (Cucumis sativus L.) flower buds and fruits. J. Appl. Genet. 2009, 50, 9–16. [Google Scholar] [CrossRef]
- Kohli, A.; González-Melendi, P.; Abranches, R.; Capell, T.; Stoger, E.; Christou, P. The quest to understand the basis and mechanisms that control expression of introduced transgenes in crop plants. Plant Signal. Behav. 2006, 1, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Cai, G.; Kong, F.; Deng, Y.; Ma, N.; Meng, Q. Overexpression of tomato chloroplast-targeted DnaJ protein enhances tolerance to drought stress and resistance to Pseudomonas solanacearum in transgenic tobacco. Plant Physiol. Biochem. 2014, 82, 95–104. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, J.; Chen, T.; Liu, Q.; Zhang, H.; Wang, Y.; Hong, Y.; Xiao, F.; Zhang, L.; Shen, Q. Type I J-domain NbMIP1 proteins are required for both Tobacco mosaic virus infection and plant innate immunity. PLoS Pathog. 2013, 9, e1003659. [Google Scholar] [CrossRef]
- Hofius, D.; Maier, A.T.; Dietrich, C.; Jungkunz, I.; Bornke, F.; Maiss, E.; Sonnewald, U. Capsid protein-mediated recruitment of host DnaJ-like proteins is required for Potato virus Y infection in tobacco plants. J. Virol. 2007, 81, 11870–11880. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.L.; Liang, S.; Wang, H.Y.; Han, L.B.; Wang, F.X.; Cheng, H.Q.; Wu, X.M.; Qu, Z.L.; Wu, J.H.; Xia, G.X. Cotton major latex protein 28 functions as a positive regulator of the ethylene responsive factor 6 in defense against Verticillium dahliae. Mol. Plant 2015, 8, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois-Meurinne, M.; Gachon, C.M.; Saindrenan, P. Pathogen-responsive expression of glycosyltransferase genes UGT73B3 and UGT73B5 is necessary for resistance to Pseudomonas syringae pv tomato in Arabidopsis. Plant Physiol. 2005, 139, 1890–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemen, A.C.; Honkanen, S.; Melton, R.E.; Findlay, K.C.; Mugford, S.T.; Hayashi, K.; Haralampidis, K.; Rosser, S.J.; Osbourn, A. Investigation of triterpene synthesis and regulation in oats reveals a role for β-amyrin in determining root epidermal cell patterning. Proc. Natl. Acad. Sci. USA 2014, 111, 8679–8684. [Google Scholar] [CrossRef] [Green Version]
- Popowich, E.A.; Firsov, A.P.; Mitiouchkina, T.Y.; Filipenya, V.L.; Dolgov, S.V.; Reshetnikov, V.N. Agrobacterium-mediated transformation of Hyacinthus orientalis with thaumatin II gene to control fungal diseases. Plant Cell Tissue Organ Cul. 2007, 90, 237–244. [Google Scholar] [CrossRef]
- Dao, T.T.H.; Linthorst, H.J.M.; Verpoorte, R. Chalcone synthase and its functions in plant resistance. Phytochem. Rev. 2011, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Kaschani, F.; Gu, C.; Niessen, S.; Hoover, H.; Cravatt, B.F.; Van der Hoorn, R.A. Diversity of serine hydrolase activities of unchallenged and botrytis-infected Arabidopsis thaliana. Mol. Cell. Proteom. 2009, 8, 1082–1093. [Google Scholar] [CrossRef] [Green Version]
- Pazouki, L.; Memari, H.R.; Kännaste, A.; Bichele, R.; Niinemets, Ü. Germacrene A synthase in yarrow (Achillea millefolium) is an enzyme with mixed substrate specificity: Gene cloning, functional characterization and expression analysis. Front. Plant Sci. 2015, 6, 111. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yin, X.; Yang, W. GDSL lipases modulate immunity through lipid homeostasis in rice. PLoS Pathog. 2017, 13, e1006724. [Google Scholar] [CrossRef] [Green Version]
- Hardham, A.R.; Jones, D.A.; Takemoto, D. Cytoskeleton and cell wall function in penetration resistance. Curr. Opin. Plant Biol. 2007, 10, 342–348. [Google Scholar] [CrossRef]
- Cantero, A.; Barthakur, S.; Bushart, T.J.; Chou, S.; Morgan, R.O.; Fernandez, M.P.; Clark, G.B.; Roux, S.J. Expression profiling of the Arabidopsis annexin gene family during germination, de-etiolation and abiotic stress. Plant Physiol. Biochem. 2006, 44, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Tian, Z.; Lu, L.; Chen, X.; Chen, X.; Zhang, W.; Song, R. Editing of mitochondrial transcripts nad3 and cox2 by Dek10 is essential for mitochondrial function and maize plant development. Genetics 2017, 205, 1489–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, Y.P. Calcium/calmodulin-dependent serine protein kinase and mental retardation. Ann. Neurol. 2009, 66, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Ouakfaoui, S.E.; Miki, B. The Stability of the Arabidopsis Transcriptome in Transgenic Plants Expressing the Marker Genes NptII and UidA. Plant J. 2005, 41, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Rai, K.M.; Srivastava, M.; Kumar, V.; Pandey, B.; Singh, S.P.; Bag, S.K.; Singh, B.D.; Tuli, R.; Sawant, S.V. Distinct role of core promoter architecture in regulation of light-mediated responses in plant genes. Mol. Plant 2014, 7, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Iwakawa, H.; Tomari, Y. Molecular Insights into microRNA-Mediated Translational Repression in Plants. Mol. Cell 2013, 52, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Nardini, M.; Gnesutta, N.; Donati, G.; Gatta, R.; Forni, C.; Fossati, A.; Vonrhein, C.; Morac, D.; Romier, C.; Blognesi, M.; et al. Sequence-specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination. Cell 2013, 152, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, A.J.; Yang, P.; Conway, A.E.; Cinghu, S.; Freudenberg, J.M.; Yellaboina, S.; Jothi, R. Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol. Cell 2014, 55, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, D.; Kong, F.; Lin, K.; Zhang, H.; Li, G. The Arabidopsis thaliana nuclear factor Y transcription factors. Front. Plant Sci. 2017, 7, 2045. [Google Scholar] [CrossRef] [Green Version]
- Mortimer, J.C.; Laohavisit, A.; Macpherson, N.; Webb, A.; Brownlee, C.; Battey, N.H.; Davies, J.M. Annexins: Multifunctional components of growth and adaptation. J. Exp. Bot. 2008, 59, 533–544. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Line | Expression | Gene ID | Description | Fold Change | FDR |
|---|---|---|---|---|---|---|
| 1 | 212 | down | G20340 | lincRNA | 4.67 | 6.74 × 10−6 |
| 2 | 212 | down | G4326 | PHB domain membrane associated | 4.49 | 1.03 × 10−8 |
| 3 | 212 | up | G14398 | Uncharacterized protein | 88.90 | 1.78 × 10−11 |
| 4 | 224 | down | G6838 | Restriction endonuclease, type II | 9.51 | 1.03 × 10−12 |
| 5 | 224 | down | G16954 | Uncharacterized protein | 4.57 | 5.92 × 10−12 |
| 6 | 224 | down | G17313 | Xyloglucan endo-transglycosylase | 2.60 | 5.40 × 10−4 |
| 7 | 224 | down | G15599 | thylakoid lumenal 17.4 kDa protein | 2.42 | 9.97 × 10−4 |
| 8 | 224 | down | G12465 | Serine threonine-protein kinase | 2.38 | 1,07 × 10−4 |
| 9 | 224 | down | G17428 | Uncharacterized protein | 2.23 | 3.87 × 10−5 |
| 10 | 224 | down | G11114 | Chaperone DnaJ-domain containing protein | 1.87 | 5.28 × 10−4 |
| 11 | 224 | down | G13243 | cystathionine gamma-synthase | 1.82 | 6.27 × 10−4 |
| 12 | 224 | up | G853 | lincRNA | 36.75 | 4.42 × 10−4 |
| 13 | 224 | up | G21621 | lipase immunity | 19.19 | 1.07 × 10−4 |
| 14 | 224 | up | G14592 | ribosomal protein L19 | 15.91 | 3.52 × 10−5 |
| 15 | 224 | up | G9998 | glycosylphosphatidylinositol-anchored lipid | 14.39 | 3.70 × 10−4 |
| 16 | 224 | up | G6441 | plastocyanin-like domain | 13.95 | 1.14 × 10−4 |
| 17 | 224 | up | G10686 | hypothetical protein | 13.68 | 8.72 × 10−4 |
| 18 | 224 | up | G10813 | protein TRANSPARENT TESTA 12-like | 13.48 | 3.87 × 10−5 |
| 19 | 224 | up | G14791 | GDSL esterase lipase | 13.28 | 3.04 × 10−4 |
| 20 | 224 | up | G149 | germacrene D synthase-like | 12.93 | 1.68 × 10−5 |
| 21 | 224 | up | G17693 | chalcone synthase | 11.44 | 6.66 × 10−6 |
| 22 | 224 | up | G3896 | Pollen proteins Ole e I like | 10.88 | 1.22 × 10−5 |
| 23 | 224 | up | G7198 | auxin-binding protein ABP19a | 10.26 | 5.17 × 10−5 |
| 24 | 224 | up | G12174 | non-specific lipid-transfer proteins | 10.17 | 1.68 × 10−5 |
| 25 | 224 | up | G19560 | beta-Amyrin | 9.21 | 6.33 × 10−4 |
| 26 | 224 | up | G12648 | Serine hydrolase (FSH1) | 8.24 | 1.28 × 10−4 |
| 27 | 224 | up | G8549 | ribonuclease 1 isoform X1 | 7.60 | 2.14 × 10−4 |
| 28 | 224 | up | G1149 | lincRNA | 7.24 | 3.74 × 10−4 |
| 29 | 224 | up | G10467 | annexin D3-like | 6.73 | 2.97 × 10−4 |
| 30 | 224 | up | G3425 | AP-2 transcritpion factor | 5.19 | 5.17 × 10−5 |
| 31 | 224 | up | G1817 | mannitol dehydrogenase-like | 4.83 | 5.45 × 10−6 |
| 32 | 224 | up | G19558 | Acyl carrier protein 4 | 3.75 | 7.48 × 10−5 |
| 33 | 224 | up | G6726 | Plasma membrane sugar-proton symporter | 3.75 | 8.62 × 10−4 |
| 34 | 224 | up | G12598 | Nuclear transcription factor Y subunit | 3.55 | 1.07 × 10−4 |
| 35 | 224 | up | G14363 | NADH-ubiquinone oxidoreductase | 3.44 | 3.87 × 10−5 |
| 36 | 224 | up | G828 | trehalose 6-phosphate to | 3.22 | 8.72 × 10−4 |
| 37 | 224 | up | G9940 | MLP-like protein | 2.65 | 4.95 × 10−4 |
| 38 | 224 | up | G6936 | trihelix transcription factor GT | 2.49 | 2.60 × 10−9 |
| 39 | 224 | up | G15513 | O-glucosyltransferase 1-like | 2.37 | 1.68 × 10−5 |
| 40 | 224 | up | G18456 | Uncharacterized protein | 2.01 | 3.91 × 10−4 |
| 41 | 224 | up | G7760 | pentatricopeptide repeat-containing | 1.83 | 1.15 × 10−4 |
| 42 | 225 | up | G10371 | lincRNA | 104.23 | 2.71 × 10−29 |
| 43 | 225 | up | G14398 | Uncharacterized protein | 34.41 | 5.89 × 10−8 |
| 44 | 225 | up | G15468 | lincRNA | 19.74 | 1.15 × 10−9 |
| 45 | 225 | up | G6182 | calcium calmodulin-dependent serine | 7.73 | 8.87 × 10−9 |
| 46 | 225 | up | G20381 | lincRNA | 5.92 | 3.02 × 10−9 |
| 47 | 225 | up | G7760 | pentatricopeptide repeat-containing protein | 1.81 | 4.48 × 10−4 |
| Line | miRNA | Type of Expression | Log2FC | p-Val | No of Targets | Targets in DEGs | Type of Changes |
|---|---|---|---|---|---|---|---|
| 212 | 320 | down | −2.09 | 5.24 × 10−5 | 20 | no | none |
| 224 | 27 | down | −2.02 | 2.27 × 10−5 | 15 | no | none |
| 224 | 42 | up | 2.08 | 6.23 × 10−6 | 22 | no | none |
| 224 | 93 | up | 2.30 | 8.70 × 10−5 | 21 | no | none |
| 224 | 123 | up | 3.55 | 3.70 × 10−11 | 25 | no | none |
| 224 | 131 | down | −2.84 | 1.11 × 10−5 | 24 | G104671 | up |
| 224 | 206 | up | 2.23 | 2.69 × 10−4 | 18 | no | none |
| 224 | 218 | down | −3.39 | 4.52 ×10−7 | 28 | G125982 | up |
| 224 | 222 | down | −3.72 | 4.96 × 10−7 | 29 | G125982 | up |
| 224 | 289 | up | 3.28 | 2.45 × 10−9 | 23 | no | none |
| 225 | 206 | up | 2.09 | 8.66 × 10−5 | 18 | no | none |
| 225 | 218 | down | −2.20 | 1.42 × 10−5 | 28 | no | none |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawełkowicz, M.E.; Skarzyńska, A.; Sroka, M.; Szwacka, M.; Pniewski, T.; Pląder, W. Effect of Transgenesis on mRNA and miRNA Profiles in Cucumber Fruits Expressing Thaumatin II. Genes 2020, 11, 334. https://doi.org/10.3390/genes11030334
Pawełkowicz ME, Skarzyńska A, Sroka M, Szwacka M, Pniewski T, Pląder W. Effect of Transgenesis on mRNA and miRNA Profiles in Cucumber Fruits Expressing Thaumatin II. Genes. 2020; 11(3):334. https://doi.org/10.3390/genes11030334
Chicago/Turabian StylePawełkowicz, Magdalena Ewa, Agnieszka Skarzyńska, Małgorzata Sroka, Maria Szwacka, Tomasz Pniewski, and Wojciech Pląder. 2020. "Effect of Transgenesis on mRNA and miRNA Profiles in Cucumber Fruits Expressing Thaumatin II" Genes 11, no. 3: 334. https://doi.org/10.3390/genes11030334
APA StylePawełkowicz, M. E., Skarzyńska, A., Sroka, M., Szwacka, M., Pniewski, T., & Pląder, W. (2020). Effect of Transgenesis on mRNA and miRNA Profiles in Cucumber Fruits Expressing Thaumatin II. Genes, 11(3), 334. https://doi.org/10.3390/genes11030334