Identification of Genetic Locus Underlying Easy Dehulling in Rice-Tartary for Easy Postharvest Processing of Tartary Buckwheat


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Sequencing
2.2. Preprocessing of Raw Sequencing Data
2.3. Variant Analysis of SNPs and Indels
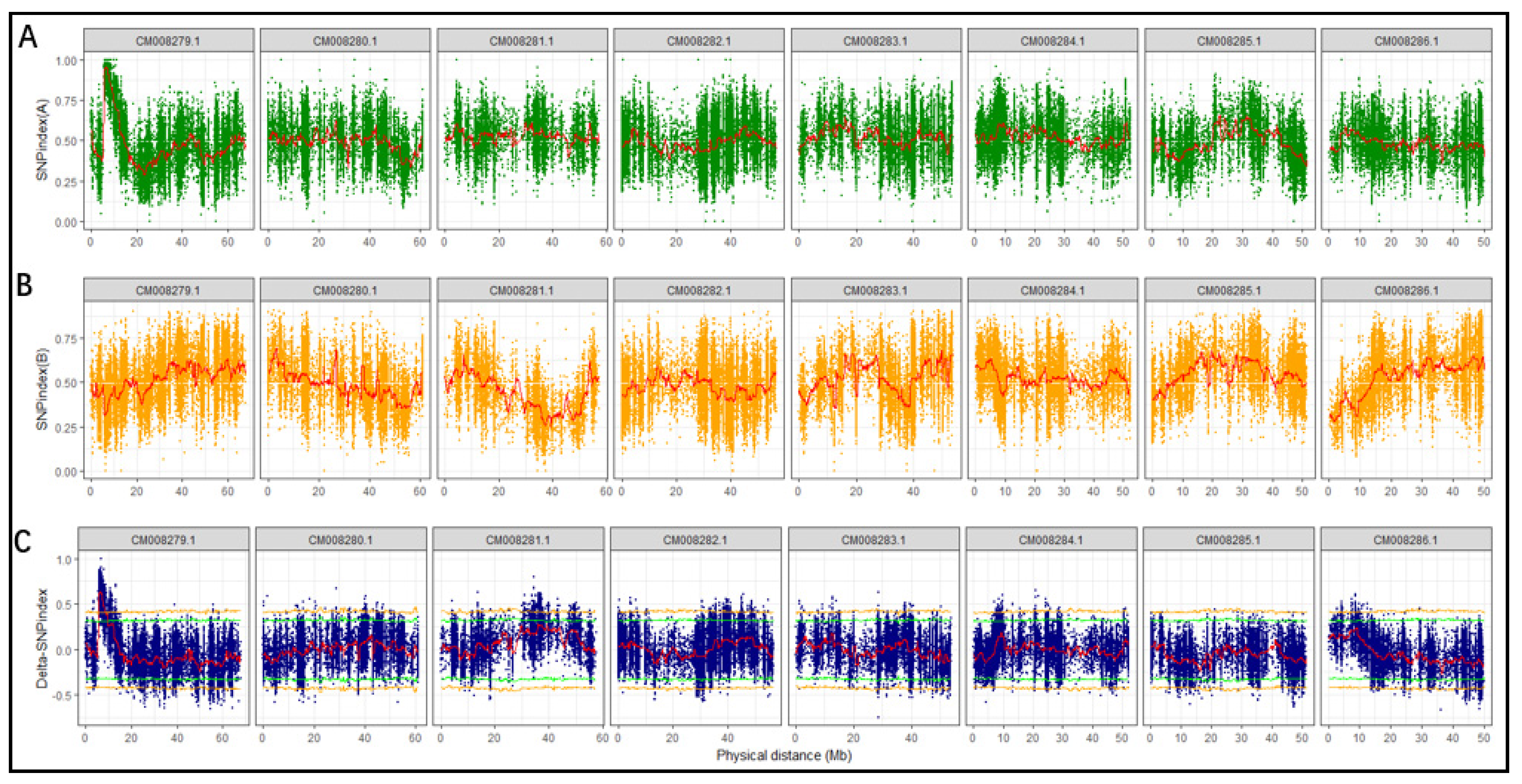
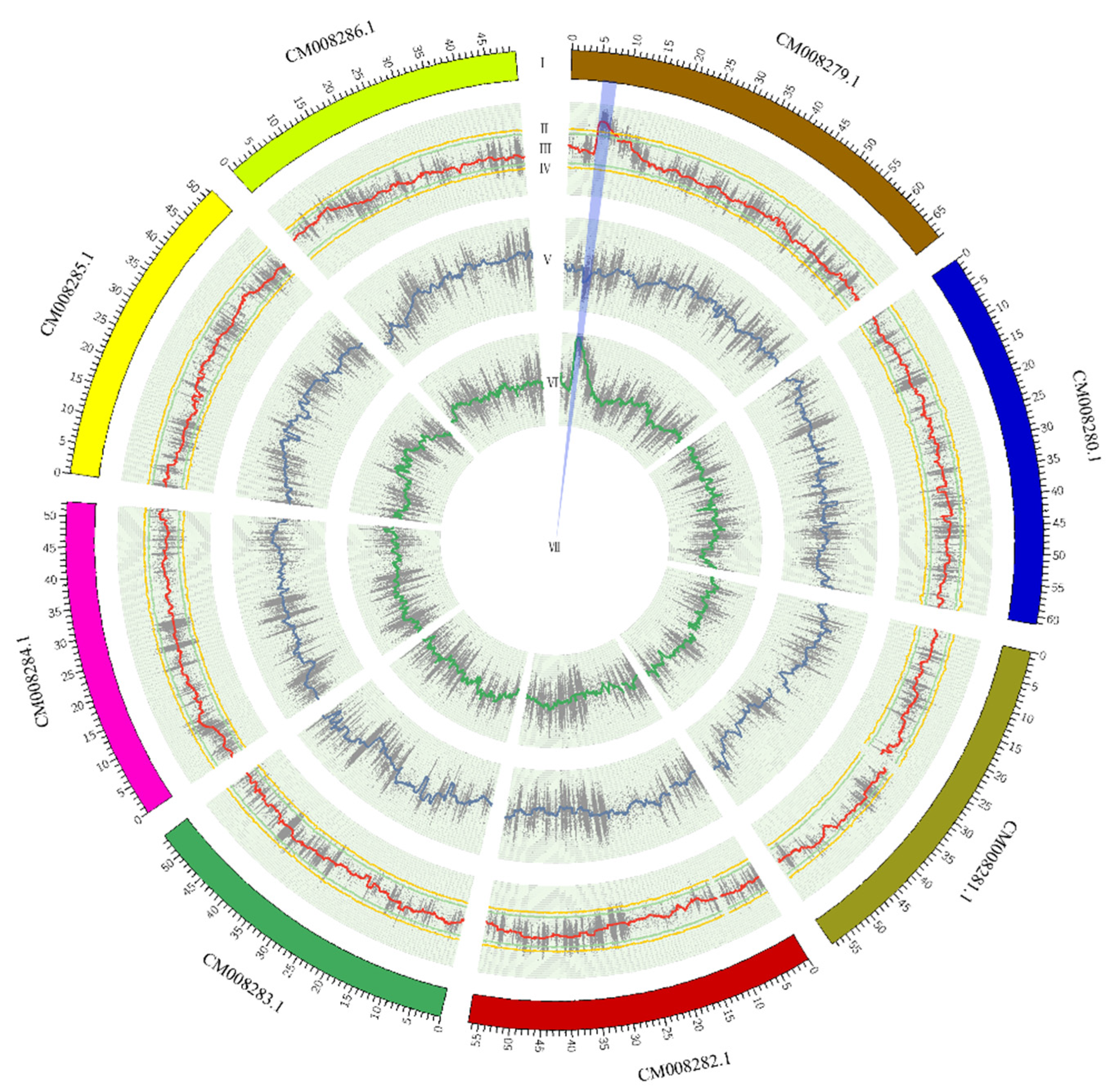
2.4. Identification of Candidate Region by Delta SNP-Index
3. Results
3.1. DNA Sequencing for Two Parents and Two Pools
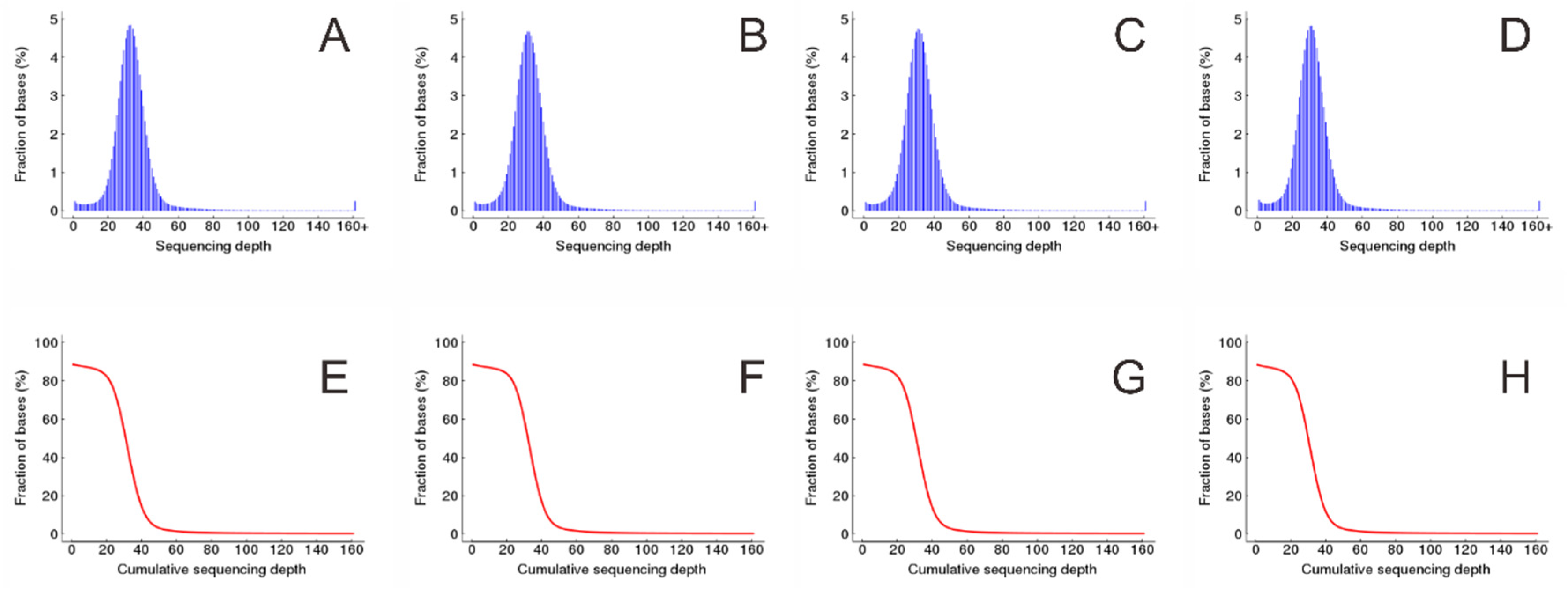
3.2. Mapping High-Quality Clean Reads to the Reference Genome
3.3. SNPs and Indels between Samples and Reference Genome
3.4. Identification of Candidate Region Underlying Easy Dehulling in Rice-Tartary according to SNP-Index
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, M.; Ivan, K.; Sun, H.W.; Nikhil, K.C.; Wieslander, G. Molecular Breeding and Nutritional Aspects of Buckwheat; Academic Press: Pittsburgh, PA, USA, 2016; pp. 203–207. [Google Scholar]
- Wang, Y.; Campbell, C.G. Tartary buckwheat breeding (Fagopyrum Tataricum L. Gaertn.) through hybridization with its Rice-Tartary type. Euphytica 2007, 156, 399–405. [Google Scholar] [CrossRef]
- Campbell, C.G. Buckwheat: Fagopyrum Esculentum Moench. Promoting the Conservation and Use of Underutilized and Neglected Crops (19); Institute of Plant Genetics and Crop Plant Research, Gatersleben, International Plant Genetic Resources Institute: Rome, Italy, 1994; 95p. [Google Scholar]
- Bonafaccia, G.; Marocchini, M.; Kreft, I. Composition and technological properties of the flour and bran from common and Tartary buckwheat. Food Chem. 2003, 80, 9–15. [Google Scholar] [CrossRef]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Suzuki, T.; Morishita, T.; Mukasa, Y.; Takigawa, S.; Yokota, S.; Ishiguro, K.; Noda, T. Breeding of ‘Manten-Kirari’, a non-bitter and trace-rutinosidase variety of Tartary buckwheat (Fagopyrum Tataricum Gaertn.). Breed. Sci. 2015, 64, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panwar, A.; Nidhi, G.; Chauhan, R. Biosynthesis and accumulation of flavonoids in Fagopyrum spp. Eur. J. Plant Sci. Biotechnol. 2012, 6, 17–26. [Google Scholar]
- Nishimura, M.; Ohkawara, T.; Sato, Y.; Satoh, H.; Suzuki, T.; Ishiguro, K.; Noda, T.; Morishita, T.; Nishihira, J. Effectiveness of Rutin-Rich Tartary buckwheat (Fagopyrum Tataricum Gaertn.) ‘Manten-Kirari’ in body weight reduction related to its antioxidant properties: A randomised, double-blind, placebo-controlled study. J. Funct. Foods 2016, 26, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Nina, F.; Rode, J.; Wang, Z.; Kreft, I.; Zhang, Z. Tartary Buckwheat (Fagopyrum Tataricum Gaertn.) as a Source of dietary rutin and quercitrin. J. Agric. Food Chem. 2003, 51, 6452–6455. [Google Scholar]
- Wang, T.Y.; Yang, K.L.; Lu, P.; Chen, W.P. Evaluation of Tartary buckwheat germplasm from Tibet and their phylogenic study. Buckwheat Trend 1996, 1, 14–21. (In Chinese) [Google Scholar]
- Hunt, H.V.; Shang, X.; Jones, M.K. Buckwheat: A crop from outside the major Chinese domestication centres? A review of the archaeobotanical, palynological and genetic evidence. Veg. Hist. Archaeobot. 2018, 27, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, O. Geographical distribution of allozymes in natural populations of wild Tartary buckwheat. Fagopyrum 2000, 17, 29–34. [Google Scholar]
- Gadaleta, A.; Colasuonno, P.; Giove, S.L.; Blanco, A.; Giancaspro, A. Map-based cloning of QFhb.mgb-2A identifies a WAK2 gene responsible for Fusarium Head Blight resistance in wheat. Sci. Rep. 2013, 126, 1839–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, L.; Zhang, Q.; Liu, Y. Map-based cloning and expression analysis of BMR-6 in sorghum. J. Genet. 2015, 94, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Huang, Q.; Sun, W.; Ma, Z.; Huang, L.; Wu, Q.; Tang, Z.; Bu, T.; Li, C.; Chen, H. Genome-wide investigation of the heat shock transcription factor (Hsf) gene family in Tartary buckwheat (Fagopyrum tataricum). BMC Genom. 2019, 20, 871. [Google Scholar] [CrossRef] [PubMed]
- Darvasi, A.; Soller, M. Selective DNA pooling for determination of linkage between a molecular marker and a quantitative trait locus. Genetics 1994, 138, 1365–1373. [Google Scholar]
- Sun, Y.; Wang, J.; Crouch, J.H.; Xu, Y. Efficiency of selective genotyping for genetic analysis of complex traits and potential applications in crop improvement. Mol. Breed. 2010, 26, 493–511. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, J.J.; Wing, R.A.; Ganal, M.W.; Tanksley, S.D. Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 1991, 19, 6553–6568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Crouch, J.H. Marker-Assisted Selection in Plant Breeding: From Publications to Practice. Crop. Sci. 2008, 48, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-Seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Macgregor, S.; Zhao, Z.Z.; Henders, A.K.; Martin, N.G.; Montgomery, G.W.; Visscher, P.M. Highly cost-efficient genome-wide association studies using DNA pools and dense SNP arrays. Nucleic Acids Res. 2008, 36, e35. [Google Scholar] [CrossRef]
- Vikram, P.; Swamy, B.P.M.; Dixit, S.; Ahmed, H.U.; Cruz, M.T.S.; Singh, A.K.; Ye, G.; Kumar, A. Bulk segregant analysis: ‘An effective approach for mapping consistent-effect drought grain yield QTLs in rice’. Field Crop. Res. 2012, 134, 185–192. [Google Scholar] [CrossRef]
- Schubert, M.; Stinus, L.; Ludovic, O. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, X.; Ma, B.; Gao, Q.; Du, H.; Han, Y.; Li, Y.; Cao, Y.; Qi, M.; Zhu, Y.; et al. The Tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol. Plant 2017, 10, 1224–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; He, L.; Li, Y.; Huang, W.; Xi, F.; Lin, L.; Zhi, Q.; Zhang, W.; Tang, Y.T.; Geng, C.; et al. OTG-Snpcaller: An optimized pipeline based on TMAP and GATK for SNP calling from Ion Torrent data. PLoS ONE 2015, 10, e0138824. [Google Scholar] [CrossRef] [Green Version]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.M.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Mingyao, L.; Hakon, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Mukasa, Y.; Suzuki, T.; Honda, Y. Suitability. of Rice-Tartary buckwheat for crossbreeding and for utilization of rutin. Jpn. Agric. Res. Q. 2009, 43, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yeh, C.-T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Matthias, J.; Kuon, J.E.; Axel, H.; Axel, A.; Sebastian, B.; Scholz, U.; Graner, A.; Stein, N. Mapping-by-sequencing accelerates forward genetics in barley. Genome Boil. 2014, 15, R78. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.W.; Hofstad, A.N.; Sreekanta, S.; Fu, F.; Kono, T.J.Y.; Orourke, J.A.; Vance, C.P.; Muehlbauer, G.J.; Stupar, R.M. Fast neutron-induced structural rearrangements at a soybean NAP1 locus result in gnarled trichomes. Theor. Appl. Genet. 2016, 129, 1725–1738. [Google Scholar] [CrossRef] [Green Version]
- Dobbels, A.A.; Michno, J.M.; Campbell, B.W.; Virdi, K.S.; Stec, A.O.; Muehlbauer, G.J.; Naeve, S.L.; Stupar, R.M. An induced chromosomal translocation in soybean disrupts a KASI ortholog and is associated with a high-sucrose and low-oil seed phenotype. Genes 2017, 7, 1215–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Lin, T.; Klein, J.; Wang, S.; Qi, J.; Zhou, Q.; Sun, J.; Zhang, Z.; Weng, Y.; Huang, S. QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor. Appl. Genet. 2014, 127, 1491–1499. [Google Scholar] [CrossRef]
- Illa-Berenguer, E.; van Houten, J.; Huang, Z.; Esther Van, D.K. Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 2015, 128, 1329–1342. [Google Scholar] [CrossRef]
- Das, S.; Upadhyaya, H.D.; Bajaj, D.; Kujur, A.; Badoni, S.; Laxmi; Kumar, V.; Tripathi, S.; Gowda, C.L.L.; Sharma, S.; et al. Deploying QTL-Seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res. 2014, 22, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, I.V. Non-Shattering Accessions of Fagopyrum tataricum Gaertn. Carry recessive alleles at two loci affecting development of functional abscission layer. Fagopyrum 2006, 23, 7–10. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean High-Quality Reads | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Read Counts | Base-Count (bp) | Reads | Bases | GC Content | Q20 Rate | Q30 Rate | |||
| Counts | Percentage | Counts (bp) | Percentage | ||||||
| KF | 175,326,806 | 26,474,347,706 | 164,150,520 | 93.63% | 23,881,470,617 | 90.21% | 37.73% | 98.77% | 95.68% |
| K21 | 167,552,432 | 25,300,417,232 | 157,245,508 | 93.85% | 22,887,256,307 | 90.46% | 37.53% | 98.79% | 95.74% |
| K5 | 165,742,386 | 25,027,100,286 | 154,379,484 | 93.14% | 22,421,430,790 | 89.59% | 37.62% | 98.74% | 95.57% |
| KM | 163,004,484 | 24,613,677,084 | 151,502,312 | 92.94% | 21,978,979,070 | 89.30% | 37.68% | 98.72% | 95.52% |
| Total | 671,626,108 | 101,415,542,308 | 624,277,824 | 91,169,136,784 | |||||
| Sample | Total Reads | All Mapped Reads | Reads with Multiple Hits | Uniquely Mapped Reads | |||
|---|---|---|---|---|---|---|---|
| Counts | Percentage | Counts | Percentages | Counts | Percentages | ||
| KF | 166,011,976 | 164,221,550 | 98.92% | 42,503,325 | 25.60% | 121,718,225 | 73.32% |
| K21 | 158,977,495 | 157,302,128 | 98.95% | 41,559,584 | 26.14% | 115,742,544 | 72.80% |
| K5 | 156,090,062 | 154,425,747 | 98.93% | 38,067,758 | 24.39% | 116,357,989 | 74.55% |
| KM | 153,020,562 | 151,421,206 | 98.95% | 39,323,846 | 25.70% | 112,097,360 | 73.26% |
| Sample | Average Depth | Percentage of Reference Bases with Different Coverage Depth | ||
|---|---|---|---|---|
| Coverage Depth ≥ 1 | Coverage Depth ≥ 4 | Coverage Depth ≥ 10 | ||
| KF | 33.29 | 88.49% | 87.90% | 86.97% |
| K21 | 32.18 | 88.57% | 88.00% | 87.00% |
| K5 | 32.12 | 88.59% | 88.04% | 87.04% |
| KM | 31.40 | 88.39% | 87.72% | 86.62% |
| Sample | SNP | Indel | ||||
|---|---|---|---|---|---|---|
| Total | Heterozygous | Homozygous | Total | Heterozygous | Homozygous | |
| KF | 356,378 | 158,972 | 197,406 | 222,008 | 22,871 | 199,137 |
| K21 | 581,386 | 457,839 | 123,547 | 261,703 | 74,637 | 187,066 |
| K5 | 582,079 | 458,763 | 123,316 | 261,989 | 74,970 | 187,019 |
| KM | 544,689 | 306,428 | 238,261 | 255,033 | 44,379 | 210,654 |
| Union | 633,256 | 543,128 | 352,990 | 270,181 | 89,724 | 230,763 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ma, M.; Liu, L. Identification of Genetic Locus Underlying Easy Dehulling in Rice-Tartary for Easy Postharvest Processing of Tartary Buckwheat. Genes 2020, 11, 459. https://doi.org/10.3390/genes11040459
Zhang L, Ma M, Liu L. Identification of Genetic Locus Underlying Easy Dehulling in Rice-Tartary for Easy Postharvest Processing of Tartary Buckwheat. Genes. 2020; 11(4):459. https://doi.org/10.3390/genes11040459
Chicago/Turabian StyleZhang, Lijun, Mingchuan Ma, and Longlong Liu. 2020. "Identification of Genetic Locus Underlying Easy Dehulling in Rice-Tartary for Easy Postharvest Processing of Tartary Buckwheat" Genes 11, no. 4: 459. https://doi.org/10.3390/genes11040459
APA StyleZhang, L., Ma, M., & Liu, L. (2020). Identification of Genetic Locus Underlying Easy Dehulling in Rice-Tartary for Easy Postharvest Processing of Tartary Buckwheat. Genes, 11(4), 459. https://doi.org/10.3390/genes11040459