Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Genotype-Recalcitrant Somatic Embryogenesis Transdifferentiation in Cotton



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Culture Conditions
2.2. Library Construction for RNA Sequencing
2.3. RNA-Seq Data and Gene Regulatory Pattern Analysis
3. Results
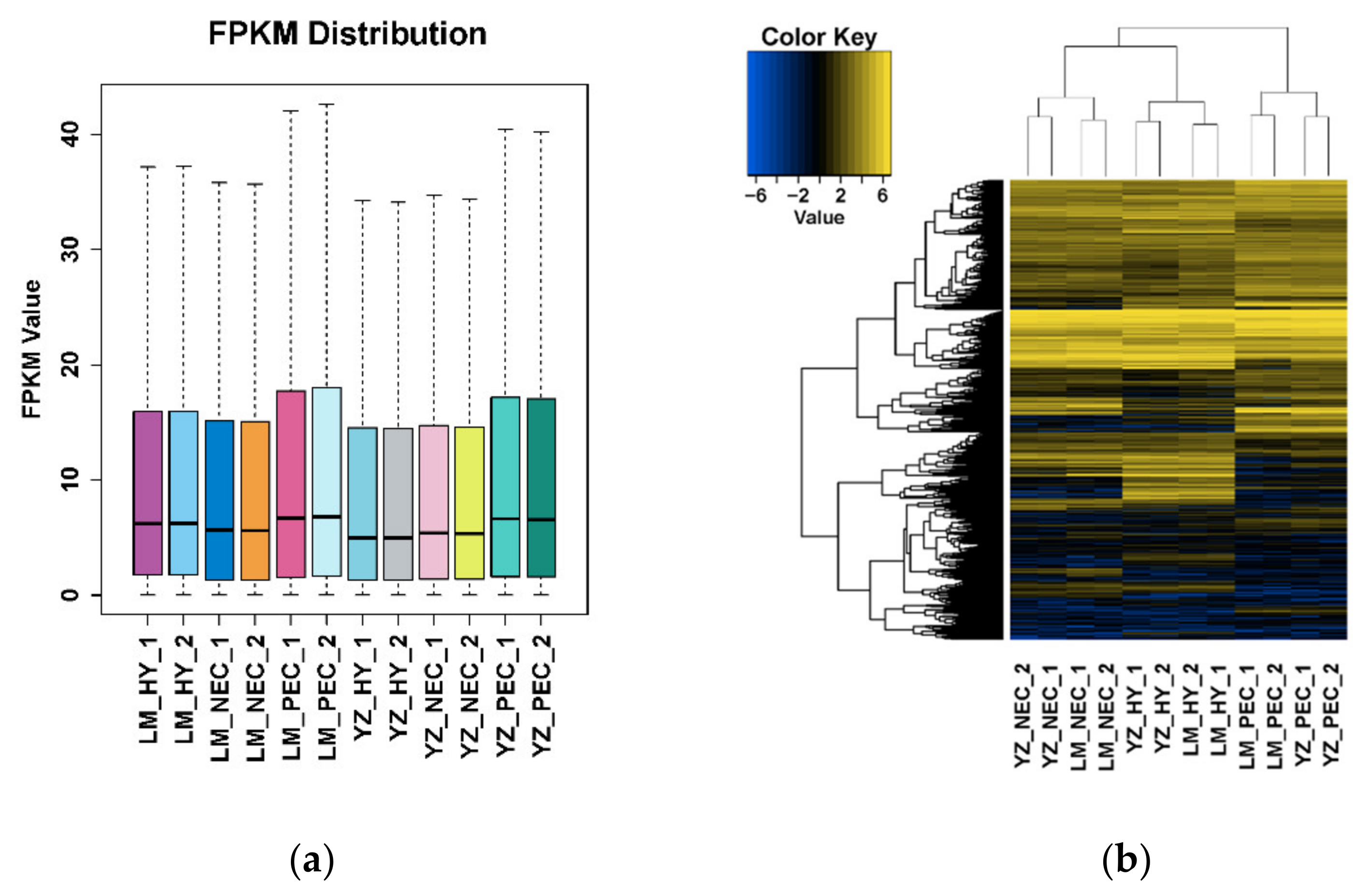
3.1. Landscape of RNA Transcriptome During Cotton SE in Two Genotypes
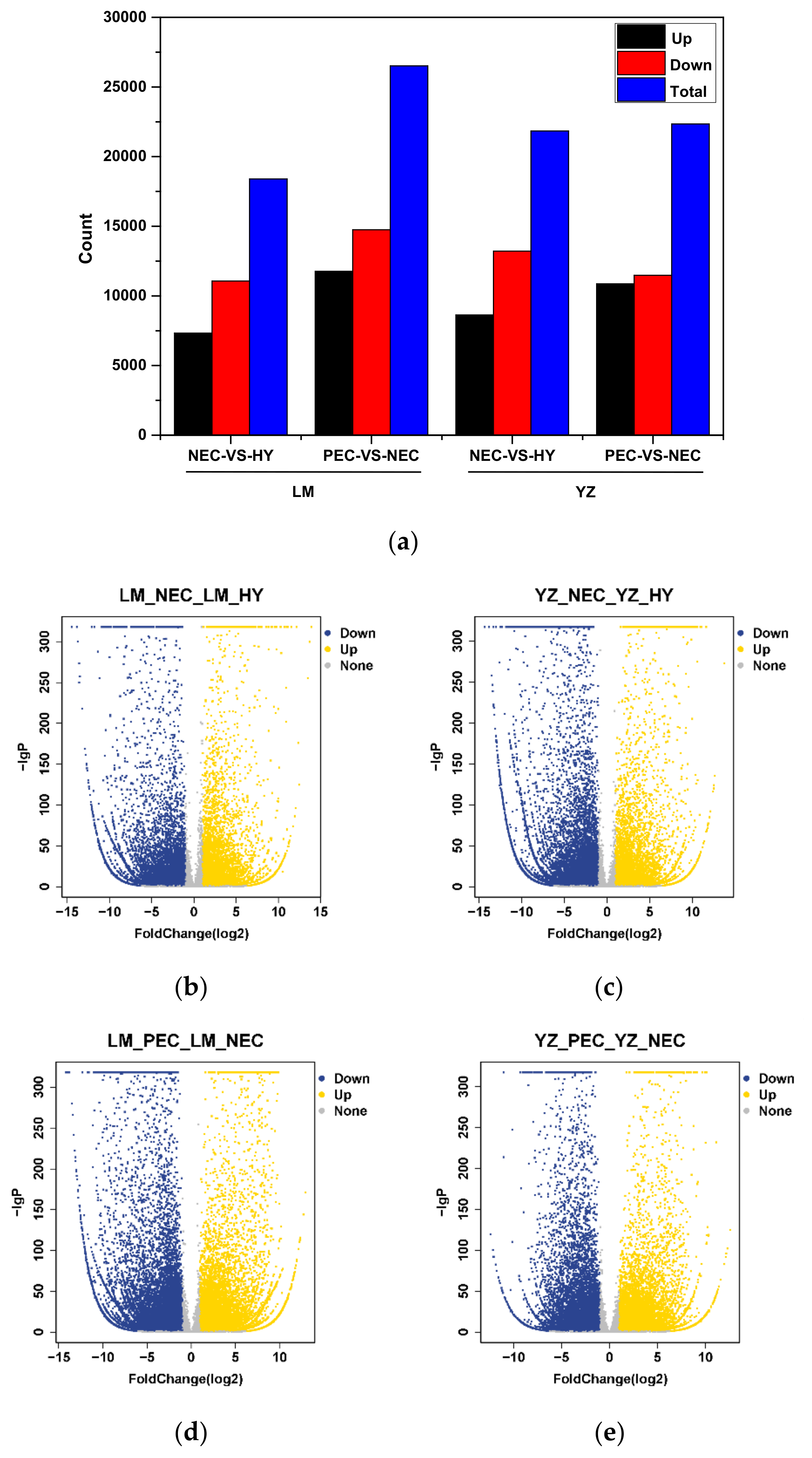
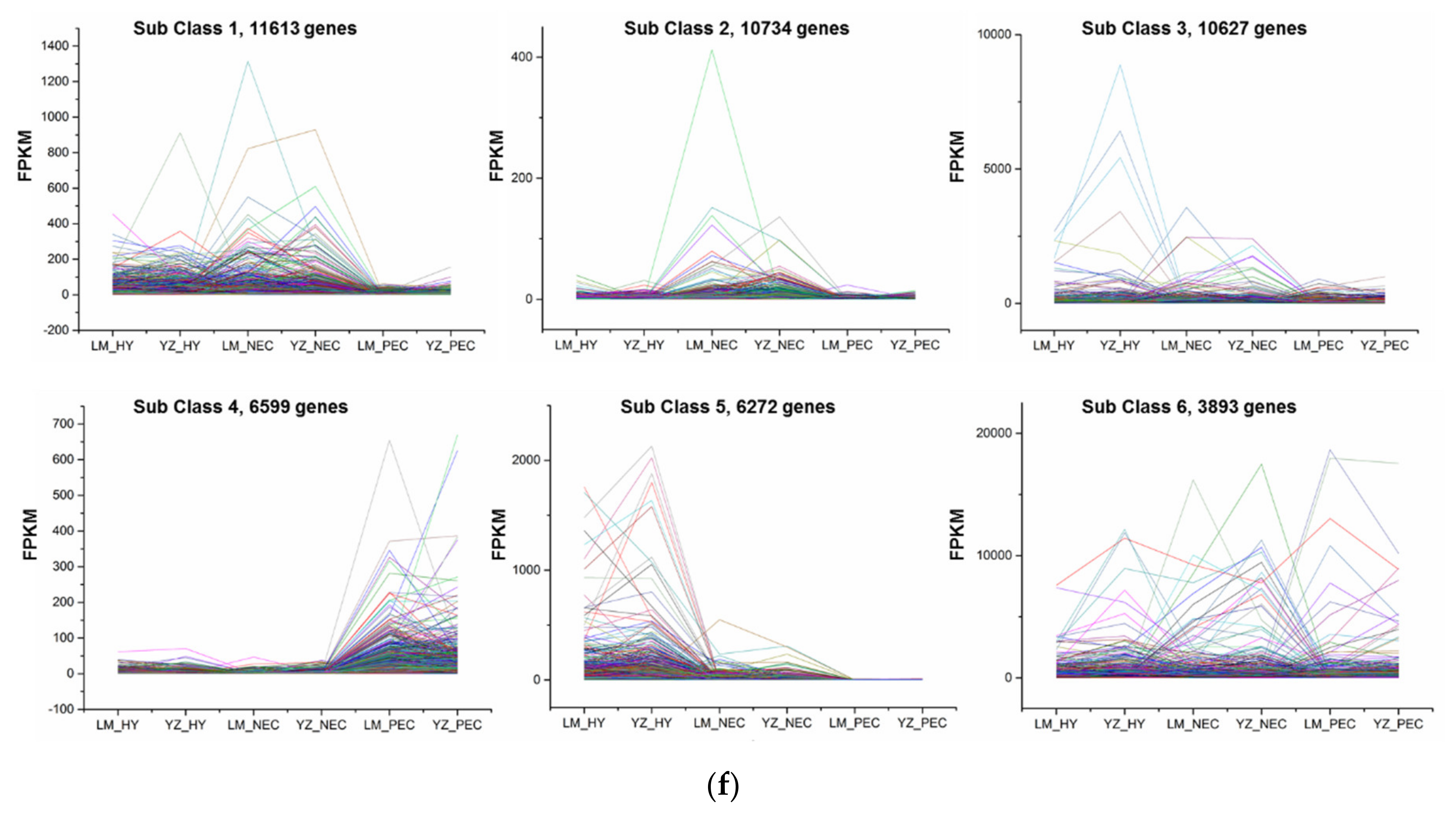
3.2. Differentially Expressed Gene Regulatory Patterns During Cotton SE
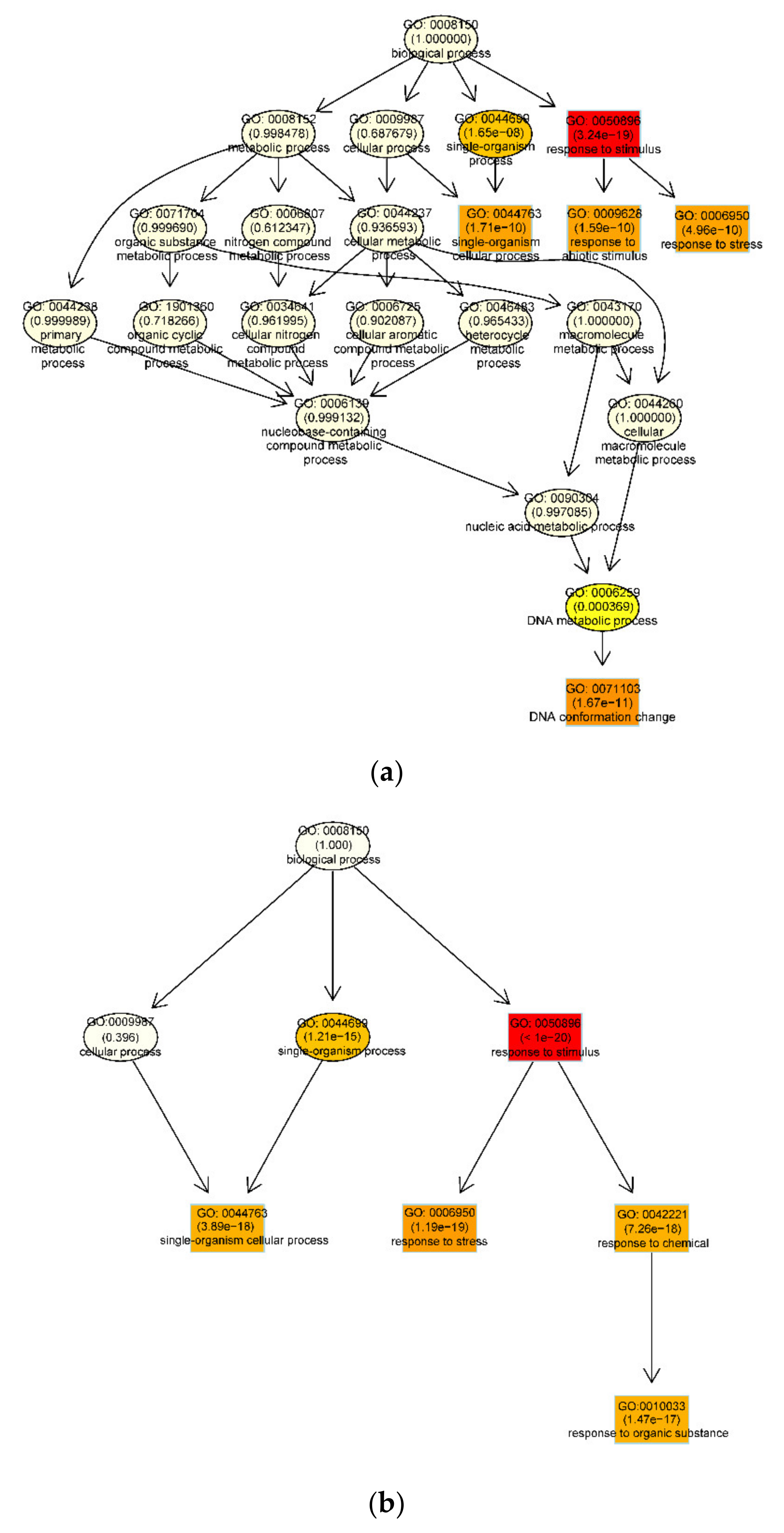
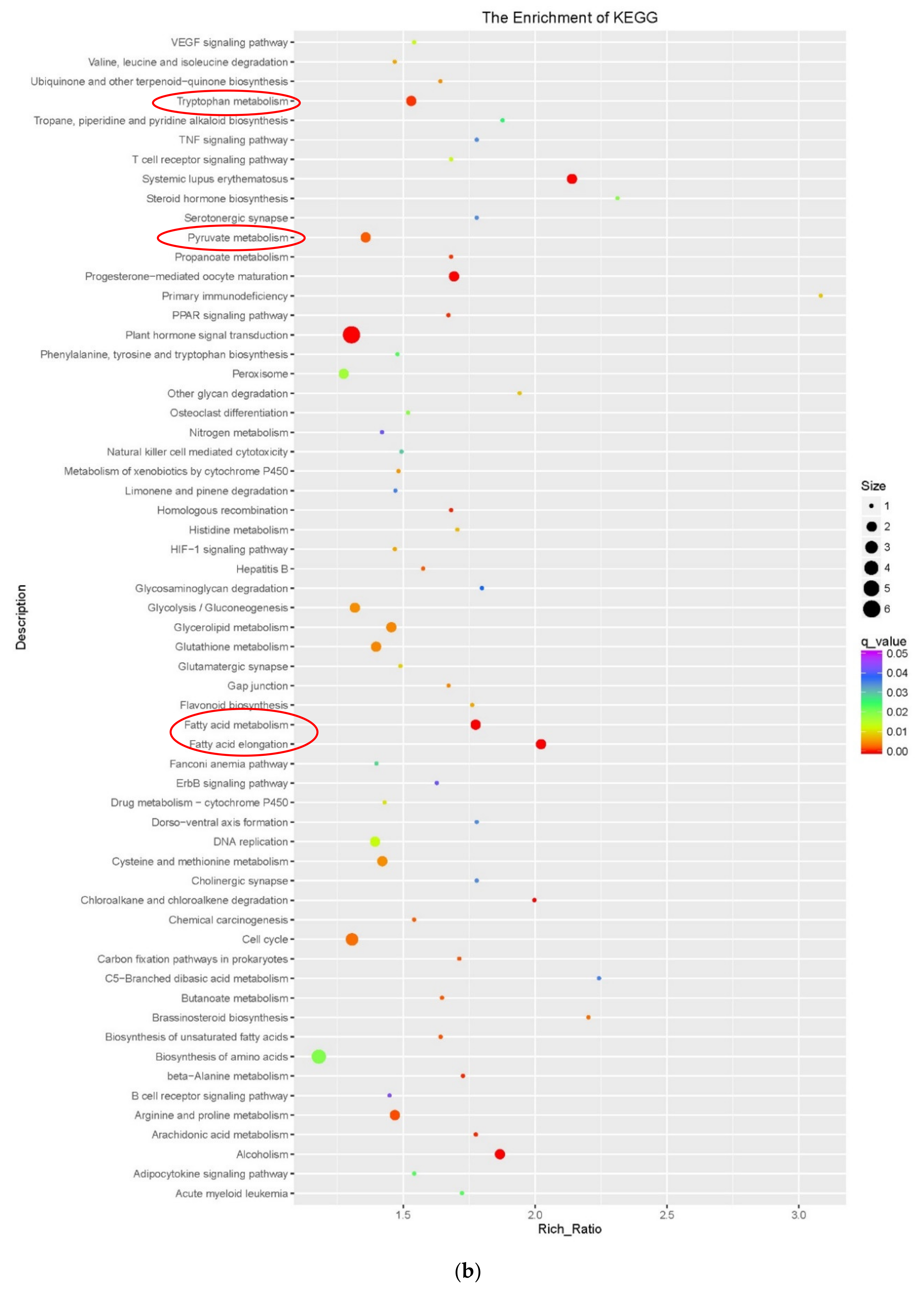
3.3. Functional Enrichment Analysis of DEGs During Embryogenic Transdifferentiation in Two Genotypes
3.3.1. DNA Conformation Change Involved in the Recalcitrant Genotype
3.3.2. Fatty Acid, Tryptophan and Pyruvate Metabolism Involved in the Highly Embryogenic Genotype
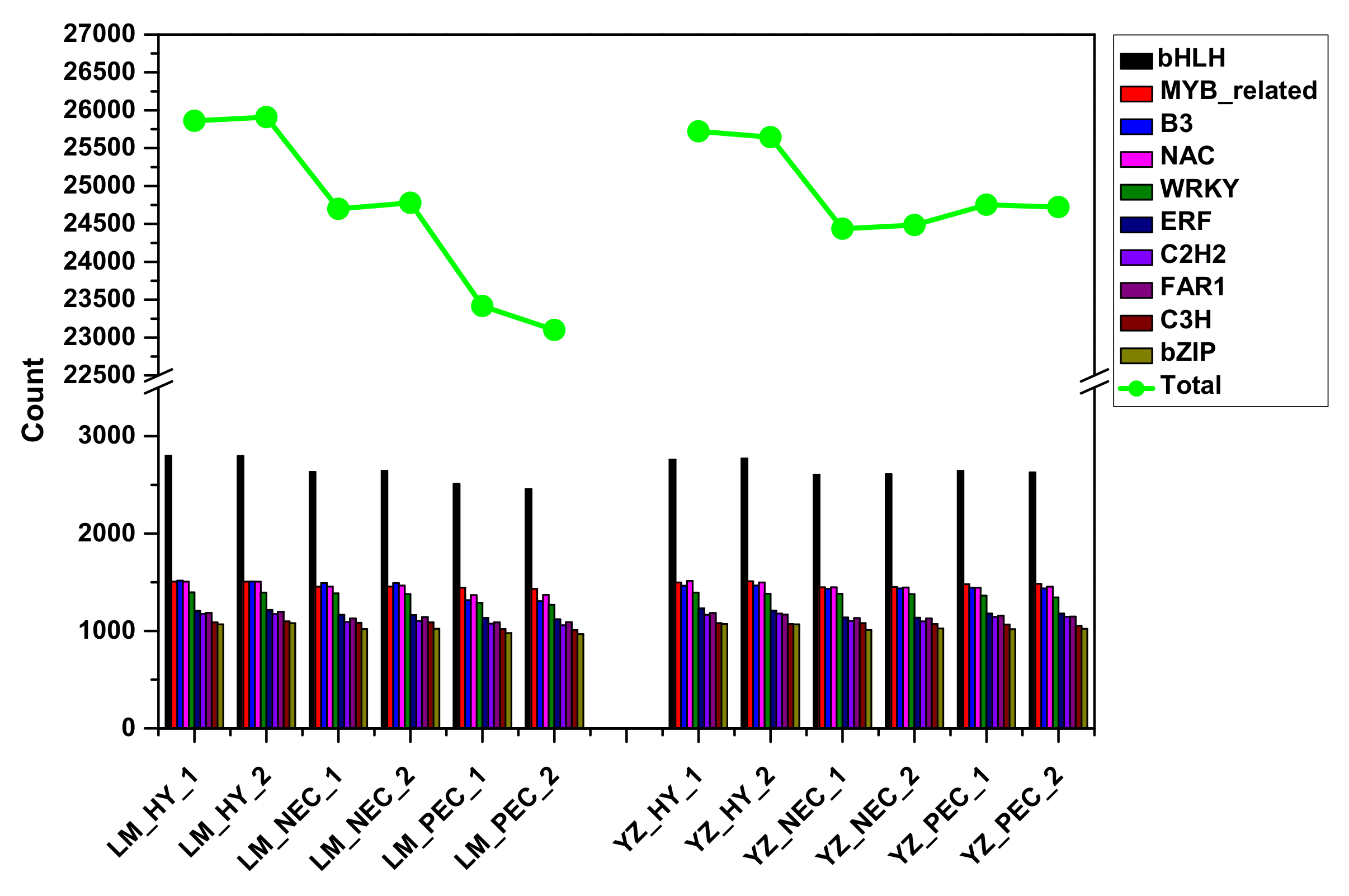
3.4. Stress-Related Transcription Factors Were Specifically Activated and Preferentially Expressed During Embryogenic Transdifferentiation in the Highly Embryogenic Genotype
3.5. Active Alternative Splicing Significantly Involved in Embryogenic Transdifferentiation in the Highly Embryogenic Genotype
4. Discussion
4.1. Effects of DNA Conformation Change During Embryogenic Transdifferentiation
4.2. Fatty Acid Metabolism Involved in Embryogenic Transdifferentiation
4.3. Pyruvate and Tryptophan Metabolism Involved in Embryogenic Transdifferentiation
4.4. Stress-Response Transcription Factors Involved in Embryogenic Transdifferentiation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A


References
- Loyola-Vargas, V.M.; De-la-Peña, C.; Galaz-Avalos, R.M.; Quiroz-Figueroa, F.R. Plant tissue culture. In Molecular Biomethods Handbook; Walker, J.M., Rapley, R., Eds.; Humana Press: Totowa, NJ, Canada, 2008; pp. 875–904. [Google Scholar]
- Yang, X.; Zhang, X. Regulation of somatic embryogenesis in higher plants. Crit. Rev. Plant Sci. 2010, 29, 36–57. [Google Scholar] [CrossRef]
- Miguel, C.; Marum, L. An epigenetic view of plant cells cultured in vitro: Somaclonal variation and beyond. J. Exp. Bot. 2011, 62, 3713–3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jopling, C.; Boue, S.; Belmonte, J. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A. Callus, Dedifferentiation, Totipotency, Somatic Embryogenesis: What These Terms Mean in the Era of Molecular Plant Biology? Front. Plant Sci. 2019, 10, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Guo, H.; Zhang, L.; Fan, Y.; Fan, Y.; Tang, Z.; Zeng, F. Dynamic TMT-based quantitative proteomics analysis of critical initiation process of totipotency during cotton somatic embryogenesis transdifferentiation. Int. J. Mol. Sci. 2019, 20, 1691–1724. [Google Scholar]
- Evans, D.A.; Sharp, W.R.; Flick, C.E. Growth and Behavior of Cell Cultures: Embryogenesis and Organogenesis. In Plant Tissue Cultures; Trevor, A.T., Ed.; Academic Press: New York, NY, USA, 1981; pp. 45–113. [Google Scholar]
- Nic-Can, G.I.; De-la-Peña, C. Epigenetic advances on somatic embryogenesis of agronomical and important crops. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications; Alvarez, V., Ed.; Springer: Berlin, Germany, 2014; pp. 91–109. [Google Scholar]
- Ikeuchi, M.; Sugimoto, K.; Iwase, A. Plant callus: Mechanisms of induction and repression. Plant Cell 2013, 25, 3159–3173. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Zhang, X.; Cheng, L. A draft gene regulatory network for cellular totipotency reprogramming during plant somatic embryogenesis. Genomics 2007, 90, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Sakhanokho, H.; Rajasekaran, K. Cotton regeneration in vitro. In Fiber Plants; Ramawat, K.G., Ahuja, M.R., Eds.; Springer: Berlin, Germany, 2016; pp. 87–110. [Google Scholar]
- Wu, J.; Zhang, X.; Nie, Y.; Jin, S.; Liang, S. Factors affecting somatic embryogenesis and plant regeneration from a range of recalcitrant genotypes of Chinese cottons (Gossypium hirsutum L.). In Vitro Cell. Dev. Plant 2004, 40, 371–375. [Google Scholar] [CrossRef]
- Zeng, F.; Zhang, X.; Zhu, L.; Tu, L.; Guo, X.; Nie, Y. Isolation and characterization of genes associated to cotton somatic embryogenesis by suppression subtractive hybridization and macroarray. Plant Mol. Biol. 2006, 60, 167–183. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, C.; Wang, Q.; Yang, Z.; Wang, Y.; Zhang, X.; Wu, Z.; Hou, Y.; Wu, J.; Li, F. iTRAQ protein profile differential analysis between somatic globular and cotyledonary embryos reveals stress, hormone, and respiration involved in increasing plantlet regeneration of Gossypium hirsutum L. J. Proteome. Res. 2014, 14, 268–278. [Google Scholar] [CrossRef]
- Cheng, W.H.; Zhu, H.G.; Tian, W.G.; Zhu, S.H.; Xiong, X.P.; Sun, Y.Q.; Zhu, Q.H.; Sun, J. De novo transcriptome analysis reveals insights into dynamic homeostasis regulation of somatic embryogenesis in upland cotton (G. hirsutum L.). Plant Mol. Biol. 2016, 92, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Cao, A.; Zheng, Y.; Yu, Y.; Wang, X.; Shao, D.; Sun, J.; Cui, B. Comparative Transcriptome Analysis of SE initial dedifferentiation in cotton of different SE capability. Sci. Rep. UK 2017, 7, 8583–8595. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha Soares, T.; da Silva, C.R.C.; Chagas Carvalho, J.M.F.; Cavalcanti, J.J.V.; de Lima, L.M.; de Albuquerque, M.F.P.; Severino, L.S.; Dos Santos, R.C. Validating a probe from GhSERK1 gene for selection of cotton genotypes with somatic embryogenic capacity. J. Biotechnol. 2018, 270, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, Y.; Ding, Y.; Wu, J.; Wang, P.; Yu, Y.; Wei, X.; Wang, Y.; Zhang, C.; Li, F.; et al. Effects of GhWUS from upland cotton (Gossypium hirsutum L.) on somatic embryogenesis and shoot regeneration. Plant Sci. 2018, 270, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.G.; Cheng, W.H.; Tian, W.G.; Li, Y.J.; Liu, F.; Xue, F.; Zhu, Q.H.; Sun, Y.Q.; Sun, J. iTRAQ-based comparative proteomic analysis provides insights into somatic embryogenesis in Gossypium hirsutum L. Plant Mol. Biol. 2018, 96, 89–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yang, X.; Li, B.; Chen, L.; Min, L.; Zhang, X. GhL1L1 affects cell fate specification by regulating GhPIN1-mediated auxin distribution. Plant Biotechnol. J. 2019, 17, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Liu, Y.; Zhou, C.; Li, X.; Zhang, X. The microRNA167 controls somatic embryogenesis in Arabidopsis through regulating its target genes ARF6 and ARF8. Plant Cell Tiss. Org. Cult. 2016, 124, 405–417. [Google Scholar] [CrossRef]
- Rupps, A.; Raschke, J.; Rümmler, M.; Linke, B.; Zoglauer, K. Identification of putative homologs of Larix decidua to BABY BOOM (BBM), LEAFY COTYLEDON1 (LEC1), WUSCHEL-related HOMEOBOX2 (WOX2) and SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE (SERK) during somatic embryogenesis. Planta 2016, 243, 473–488. [Google Scholar] [CrossRef]
- Elhiti, M.; Tahir, M.; Gulden, R.; Khamiss, K.; Stasolla, C. Modulation of embryo-forming capacity in culture through the expression of Brassica genes involved in the regulation of the shoot apical meristem. J. Exp. Bot. 2010, 61, 4069–4085. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Xiong, L.; Yang, Y. Rice SERK1 gene positively regulates somatic embryogenesis of cultured cell and host defense response against fungal infection. Planta 2005, 222, 107–117. [Google Scholar] [CrossRef]
- Kumar, V.; Van Staden, J. New insights into plant somatic embryogenesis: An epigenetic view. Acta Physiol. Plant 2017, 39, 194–211. [Google Scholar] [CrossRef]
- Hu, L.; Yang, X.; Yuan, D.; Zeng, F.; Zhang, X. GhHmgB3 deficiency deregulates proliferation and differentiation of cells during somatic embryogenesis in cotton. Plant Biotechnol. J. 2011, 9, 1038–1048. [Google Scholar] [CrossRef]
- Poon, S.; Heath, R.; Clarke, A. A chimeric arabinogalactan protein promotes somatic embryogenesis in cotton cell culture. Plant Physiol. 2012, 160, 684–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, L.; Hu, Q.; Li, Y.; Xu, J.; Ma, Y.; Zhu, L.; Yang, X.; Zhang, X. Leafy cotyledon1-casein kinase I-TCP15-phytochrome interacting factor 4 network regulates somatic embryogenesis by regulating auxin homeostasis. Plant Physiol. 2015, 169, 2805–2821. [Google Scholar] [PubMed] [Green Version]
- Jin, F.; Hu, L.; Yuan, D.; Xu, J.; Gao, W.; He, L.; Yang, X.; Zhang, X. Comparative transcriptome analysis between somatic embryos (SEs) and zygotic embryos in cotton: Evidence for stress response functions in SE development. Plant Biotechnol. J. 2014, 12, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Yuan, D.; Jin, F.; Zhang, Y.; Xu, J. Transcript profiling reveals complex auxin signaling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biol. 2012, 12, 110–128. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhang, C.; Zhang, X.; Liu, C.; Wu, Z.; Yang, Z.; Zhou, K.; Yang, X.; Li, F. Transcriptome profiling reveals auxin and cytokinin regulating somatic embryogenesis in different sister lines of cotton cultivar CCRI24. J. Integr. Plant Biol. 2013, 55, 631–642. [Google Scholar] [CrossRef]
- Kennedy, D.; Norman, C. What don’t we know? Science 2005, 309, 75. [Google Scholar] [CrossRef] [Green Version]
- Vogel, G. How does a single somatic cell become a whole plant? Science 2005, 309, 86. [Google Scholar] [CrossRef] [Green Version]
- Kaeppler, S.M.; Kaeppler, H.F.; Rhee, Y. Epigenetic aspects of somaclonal variation in plants. Plant Mol. Biol. 2000, 43, 179–188. [Google Scholar] [CrossRef]
- De-la-Peña, C.; Nic-Can, G.I.; Galaz-Ávalos, R.M.; Avilez-Montalvo, R.; Loyola-Vargas, V.M. The role of chromatin modifications in somatic embryogenesis in plants. Front. Plant Sci. 2015, 6, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Yu, X.; Guo, H.; Wei, J.; Guo, H.; Zhang, L.; Zeng, F. Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Dose IBA-Induced Embryogenic Redifferentiation in Cotton. Int. J. Mol. Sci. 2020, 21, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, D.; Jin, S.; Guo, X.; Zhang, X. Somatic Embryogenesis and Plant Regeneration in Cotton Cultivars from Yellow and Yangtze River Planting Areas. Acta Agron. Sin. 2007, 33, 394–400. [Google Scholar]
- Feng, R.; Wang, Q.; Zhang, B.; Yu, X. Genotype analysis in cotton tissue culture and plant regeneration. Acta Agric. Boreali Occident. Sin. 1997, 6, 27–30. [Google Scholar]
- Zhang, C.; Yu, S.; Fan, S.; Zhang, J.; Li, F. Inheritance of somatic embryogenesis using leaf petioles as explants in upland cotton. Euphytica 2011, 181, 55–63. [Google Scholar] [CrossRef]
- Zhang, X.L.; Sun, J.Z.; Liu, J.L. Somatic embryogenesis and plant regeneration in upland cotton. Acta Genet. Sin. 1991, 18, 461–467. [Google Scholar]
- Mishra, R.; Wang, H.Y.; Yadav, N.R.; Wilkins, T.A. Development of a highly regenerable elite Acala cotton (Gossypium hirsutum cv. Maxxa): A step towards genotype-independent regeneration. Plant Cell Tiss. Org. 2003, 73, 21–35. [Google Scholar] [CrossRef]
- Ganesan, M.; Jayabalan, N. Evaluation of haemoglobin (erythrogen): For improved somatic embryogenesis and plant regeneration in cotton (Gossypium hirsutum L. cv. SVPR 2). Plant Cell Rep. 2004, 23, 181–187. [Google Scholar] [CrossRef]
- Guo, H.; Guo, H.; Zhang, L.; Fan, Y.; Fan, Y.; Zeng, F. SELTP assembled battery drives totipotency of somatic plant cell. Plant Biotechnol. J. 2019, 17, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Wu, J.; Chen, C.; Wang, H.; Zhao, Y.; Zhang, C.; Jia, Y.; Liu, F.; Ning, T.; Chu, Z.; et al. Identification and characterization of cell cultures with various embryogenic/regenerative potential in cotton based on morphological, cytochemical, and cytogenetical assessment. J. Integr. Agr. 2019, 18, 1–8. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, 293–297. [Google Scholar] [CrossRef]
- Guo, H.; Fan, Y.; Guo, H.; Wu, J.; Yu, X.; Wei, J.; Lian, X.; Zhang, L.; Gou, Z.; Fan, Y.; et al. Somatic embryogenesis critical initiation stage-specific mCHH hypomethylation reveals epigenetic basis underlying embryogenic redifferentiation in cotton. Plant Biotechnol. J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Iyerpascuzzi, A.; Jackson, T.; Cui, H.; Petricka1, J.; Busch, W.; Tsukagoshi, H.; Benfey, P. Cell identity regulators link development and stress responses in the Arabidopsis root. Dev. Cell 2011, 21, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188–211. [Google Scholar] [CrossRef]
- Duarte-Aké, F.; De-la-Peña, C. Epigenetic advances in somatic embryogenesis in sequenced genome crops. In Somatic Embryogenesis: Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer: Berlin, Germany, 2016; pp. 81–102. [Google Scholar]
- Loyola-Vargas, V.M.; Ochoa-Alejo, N. An introduction to plant cell culture: The future ahead. In Plant Cell Culture Protocols; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Humana Press: Totowa, Canada, 2012; pp. 1–8. [Google Scholar]
- Feuerstein, B.G.; Pattabiraman, N.; Marton, L.J. Molecular mechanics of the interactions of spermine with DNA: DNA bending as a result of ligand binding. Nucleic Acids Res. 1990, 18, 1271–1282. [Google Scholar] [CrossRef]
- Salo, H.M.; Sarjala, T.; Jokela, A.; Häggman, H.; Vuosku, J. Moderate stress responses and specific changes in polyamine metabolism characterize Scots pine somatic embryogenesis. Tree Physiol. 2016, 36, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Bravo, S.; Bertín, A.; Turner, A.; Sepúlveda, F.; Jopia, P.; Parra, M.J.; Castillo, R.; Hasbún, R. Differences in DNA methylation, DNA structure and embryogenesis-related gene expression between embryogenic and non embryogenic lines of Pinus radiata D. don. Plant Cell Tiss. Org. 2017, 130, 521–529. [Google Scholar] [CrossRef]
- Corredoira, E.; Cano, V.; Bárány, I.; Solís, M.T.; Rodríguez, H.; Viéitez, M.; Ana, M.; Risueño, M.C.; Testillano, P.S. Initiation of leaf somatic embryogenesis involves high pectin esterification, auxin accumulation and DNA demethylation in Quercus alba. J. Plant Physiol. 2017, 213, 42–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnholdt-Schmitt, B. Stress-induced cell reprogramming. A role for global genome regulation? Plant Physiol. 2004, 136, 2579–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarty, D.; Yu, K.; Paek, K. Detection of DNA methylation changes during somatic embryogenesis of Siberian ginseng (Eleuterococcus senticosus). Plant Sci. 2003, 165, 61–68. [Google Scholar] [CrossRef]
- Noceda, C.; Salaj, T.; Pérez, M.; Viejo, M.; Cañal, J.; Salaj, J.; Rodriguez, R. DNA demethylation and decrease on free polyamines is associated with the embryogenic capacity of Pinus nigra Arn. cell culture. Trees 2009, 23, 1285–1293. [Google Scholar] [CrossRef]
- Viejo, M.; Rodríguez, R.; Valledor, L.; Pérez, M.; Cañal, M.; Hasbún, R. DNA methylation during sexual embryogenesis and implications on the induction of somatic embryogenesis in Castanea sativa Miller. Sex. Plant Reprod. 2010, 23, 315–323. [Google Scholar] [CrossRef]
- Nic-Can, G.; Lopez-Torres, A.; Barredo-Pool, F.; Wrobel, K.; Loyola-Vargas, V.; Rojas-Herrera, R.; De-la-Peña, C. New insights into somatic embryogenesis: LEAFY COTYLEDON1, BABY BOOM1 and WUSCHEL-RELATED HOMEOBOX4 are epigenetically regulated in Coffea canephora. PLoS ONE 2013, 8, e72160. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Liu, C.; Xia, H.; Bi, Y.; Zhao, C.; Zhao, S.; Hou, L.; Li, F.; Wang, X. Induced expression of AtLEC1 and AtLEC2 differentially promotes somatic embryogenesis in transgenic tobacco plants. PLoS ONE 2013, 8, e71714. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Tan, H.; Zheng, Q.; Fu, F.; Liang, Y.; Zhang, J.; Yang, X.; Wang, T.; Chong, K.; Wang, X.J.; et al. LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in Arabidopsis. Plant Physiol. 2008, 148, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.S.; Dubreucq, B.; Miquel, M.; Caboche, M.; Lepiniec, L. LEAFY COTYLEDON 2 activation is sufficient to trigger the accumulation of oil and seed specific mRNAs in Arabidopsis leaves. FEBS Lett. 2005, 579, 4666–4670. [Google Scholar] [CrossRef] [Green Version]
- Domżalska, L.; Kędracka-Krok, S.; Jankowska, U.; Grzyb, M.; Sobczak, M.; Rybczyński, R.J.J.; Mikuła, A. Proteomic analysis of stipe explants reveals differentially expressed proteins involved in early direct somatic embryogenesis of the tree fern Cyathea delgadii Sternb. Plant Sci. 2017, 258, 61–76. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, P.; Slabas, A.R.; Fawcett, T. Fatty acid and lipid biosynthetic genes are expressed at constant molar ratios but different absolute levels during embryogenesis. Plant Physiol. 2002, 129, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Garay, A.; Lopez, J.A.; Camafeita, E.; Bueno, M.A.; Pintos, B. Proteomic perspective of Quercus suber somatic embryogenesis. J. Proteom. 2013, 93, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, S.P.; Shmakov, V.N.; Dudareva, L.V.; Stolbikova, A.V.; Semenova, N.V.; Tret’yakova, I.N.; Konstantinov, Y.M. Fatty acid composition of total lipids in embryogenic and nonembryogenic callus lines of larch. Russ. J. Plant Physiol. 2016, 63, 252–258. [Google Scholar] [CrossRef]
- Molino, D.; Markham, J.; Bellec, Y.; Gissot, L.; Palauqui, J.; Moreau, P.; Napier, J.; Faure, J. Very long chain fatty acids are required for cell polarity and organogenesis during plant development. Chem. Phys. Lipids 2010, 163, S17. [Google Scholar] [CrossRef]
- MacNicol, P.; Jacobsen, J. Regulation of alcohol dehydrogenase gene expression in barley aleurone by giberellin and abscisic acid. Physiol. Plant. 2001, 111, 533–539. [Google Scholar] [CrossRef]
- Altamura, M.M.; Della Rovere, F.; Fattorini, L.; D’Angeli, S.; Falasca, G. Recent advances on genetic and physiological bases of in vitro somatic embryo formation. In Vitro Embryogenesis in Higher Plants; Humana Press: New York, NY, USA, 2016; pp. 47–85. [Google Scholar]
- Wójcikowska, B.; Jaskóla, K.; Gąsiorek, P.; Meus, M.; Nowak, K.; Gaj, M.D. LEAFY COTYLEDON2 (LEC2) promotes embryogenic induction in somatic tissues of Arabidopsis, via YUCCA -mediated auxin biosynthesis. Planta 2013, 238, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, I.; Thapaliya, M.; Boroujerdi, A.; Chowdhury, K. NMR-based metabolomics study of the biochemical relationship between sugarcane callus tissues and their respective nutrient culture media. Anal. Bioanal. Chem. 2014, 406, 5997–6005. [Google Scholar] [CrossRef]
- Shen, Y.; Jiang, Z.; Yao, X.; Zhang, Z.; Lin, H.; Zhao, M.; Liu, H.; Peng, H.; Li, S.; Pan, G. Genome expression profile analysis of the immature maize embryo during dedifferentiation. PLoS ONE 2012, 7, e32237. [Google Scholar] [CrossRef] [Green Version]
- Ayil-Gutiérrez, B.; Galaz-Ávalos, R.; Peña-Cabrera, E.; Loyola-Vargas, V. Dynamics of the concentration of IAA and some of its conjugates during the induction of somatic embryogenesis in Coffea canephora. Plant Signal. Behav. 2013, 8, e26998. [Google Scholar] [CrossRef] [Green Version]
- Kumaravel, M.; Uma, S.; Backiyarani, S.; Saraswathi, M.; Vaganan, M.; Muthusamy, M.; Sajith, K. Differential proteome analysis during early somatic embryogenesis in Musa spp. AAA cv. Grand Naine. Plant Cell Rep. 2016, 36, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Noaha, A.; Niemenaka, N.; Sunderhausb, S.; Haaseb, C.; Omokoloa, D.; Winkelmannc, T.; Braunb, H. Comparative proteomic analysis of early somatic and zygotic embryogenesis in Theobroma cacao L. J. Proteom. 2013, 78, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Tognetti, V.; Mühlenbock, P.; Van Breusegem, B. Stress homeostasis-the redox and auxin perspective. Plant Cell Environ. 2012, 35, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Grafi, G.; Barak, S. Stress induces cell dedifferentiation in plants. Biochim. Biophys. Acta 2015, 1849, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A. Somatic embryogenesis-stress induced remodeling of plant cell fate. Biochim. Biophys. Acta 2015, 1849, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Guisez, Y. Stress-induced morphogenic responses: Growing out of trouble? Trends Plant Sci. 2007, 12, 98–105. [Google Scholar]
- Fehér, A.; Pasternak, T.; Dudits, D. Transition of somatic plant cells to an embryogenic state. Plant Cell Tiss. Org. 2003, 74, 201–228. [Google Scholar] [CrossRef]
- Namasivayam, P. Acquisition of embryogenic competence during somatic embryogenesis. Plant Cell Tiss. Org. 2007, 90, 1–8. [Google Scholar] [CrossRef]
- Zhou, T.; Yang, X.; Guo, K.; Deng, J.; Xu, J.; Gao, W.; Lindsey, K.; Zhang, X. ROS homeostasis regulates somatic embryogenesis via the regulation of auxin signaling in cotton. Mol. Cell. Proteom. 2016, 15, 2108–2124. [Google Scholar] [CrossRef] [Green Version]
- Gliwicka, M.; Nowak, K.; Balazadeh, S.; Mueller-Roeber, B.; Gaj, M. Extensive modulation of the transcription factor transcriptome during somatic embryogenesis in Arabidopsis thaliana. PLoS ONE 2013, 8, e69261. [Google Scholar] [CrossRef] [Green Version]
- Iwase, A.; Harashima, H.; Ikeuchi, M.; Rymen, B.; Ohnuma, M.; Komaki, S.; Morohashi, K.; Kurata, T.; Nakata, M.; Ohme-Takagi, M.; et al. WIND1 promotes shoot regeneration through transcriptional activation of ENHANCER OF SHOOT REGENERATION1 in Arabidopsis. Plant Cell 2017, 29, 54–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhiti, M.; Stasolla, C.; Wang, A. Molecular regulation of plant somatic embryogenesis. In Vitro Cell. Dev. Plant 2013, 49, 631–642. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Description | Log2 (Fold Change) | |
|---|---|---|---|---|
| LM_PEC/ LM_NEC | YZ_PEC/ YZ_NEC | |||
| Gh_A11G2024 | IGHMBP2 | DNA-binding protein SMUBP-2 | −1.86 | — |
| Gh_D09G2407 | SRS2 | ATP-dependent DNA helicase SRS2-like protein | −1.65 | — |
| Gh_D11G1733 | RECQL3 | ATP-dependent DNA helicase Q-like 3 | −6.37 | −4.37 |
| Gh_A05G2891 | SYN1 | Sister chromatid cohesion 1 protein 1 | −3.24 | — |
| Gh_A07G0113 | NAP1 | Nucleosome assembly protein 1 | −1.01 | — |
| Gh_A09G0152 | SMC4 | Structural maintenance of chromosomes protein 4 | −4.98 | 2.74 |
| Gh_A08G2038 | CENPV | Centromere protein V | −2.63 | −1.10 |
| Gh_A05G2192 | TFG2 | Transcription initiation factor IIF subunit beta | −1.43 | — |
| Gh_A05G3742 | CSP1 | Cold shock protein 1 | −6.26 | — |
| Gh_A07G0517 | PHRF1 | PHD and RING finger domain-containing protein 1 | −1.57 | — |
| Gh_A12G1650 | ASCC3 | Activating signal cointegrator 1 complex subunit 3 | −1.02 | — |
| Gh_D07G2021 | CDC45 | Cell division control protein 45 homolog | −2.74 | — |
| Gh_A01G1890 | HIS4 | Histone H4 | 5.07 | 3.56 |
| Gh_A02G1361 | HIS4 | Histone H4 | 4.55 | 2.42 |
| Gh_A03G0342 | HIS4 | Histone H4 | 5.36 | 3.23 |
| Gh_A08G2531 | HIS4 | Histone H4 | 3.61 | 1.72 |
| Gh_A08G2548 | HIS4 | Histone H4 | 4.92 | 2.95 |
| Gh_A10G0449 | HIS4 | Histone H4 | 4.17 | 2.23 |
| Gh_A02G0490 | CAPH2 | Condensin-2 complex subunit H2 | 3.17 | 2.29 |
| Gh_A02G1767 | GYRB | DNA gyrase subunit B, chloroplastic/mitochondrial | 2.10 | - |
| Gh_A03G0513 | SMC2-1 | Structural maintenance of chromosomes protein 2-1 | 4.01 | 2.17 |
| Gh_A03G0582 | NRP2 | NAP1-related protein 2 | 1.99 | 1.55 |
| Gh_A06G1021 | ZRANB3 | DNA annealing helicase and endonuclease | 1.10 | - |
| Gh_A03G0737 | MCM2 | DNA replication licensing factor | 2.45 | 1.70 |
| Gh_A09G0025 | MCM2 | DNA replication licensing factor | 4.83 | 3.86 |
| Gh_A05G1256 | MCM3 | DNA replication licensing factor | 3.44 | - |
| Gh_A03G1940 | MCM6 | DNA replication licensing factor | 5.18 | 2.27 |
| Gh_A04G1267 | HMGIY2 | HMG-Y-related protein A | 1.69 | - |
| Gh_A07G2178 | HMGIY2 | HMG-Y-related protein A | 2.60 | 1.51 |
| Gh_A05G2910 | DNA2 | DNA replication ATP-dependent helicase/nuclease | 3.85 | 2.54 |
| Gh_A07G0948 | IDM1 | Increased DNA methylation 1 | 6.01 | 4.99 |
| Gh_A07G1821 | SNA41 | Cell division control protein 45 homolog | 6.21 | 2.52 |
| Gh_A09G0431 | FKBP53 | Peptidyl-prolyl cis-trans isomerase | 1.87 | - |
| Gh_A09G1577 | ORTH2 | E3 ubiquitin-protein ligase ORTHRUS 2 | 2.37 | 2.67 |
| Gene ID | Gene Name | Description | Pathway Annotation | Log2 (Fold Change) | |
|---|---|---|---|---|---|
| LM_PEC/ LM_NEC | YZ_PEC/ YZ_NEC | ||||
| Gh_D02G0833 | ECHS1 | Probable enoyl-CoA hydratase, mitochondrial | Fatty acid elongation | −1.65 | −1.29 |
| Gh_A12G1958 | PPT3 | Palmitoyl-protein thioesterase 3 | Fatty acid elongation | - | 2.69 |
| Gh_A08G1244 | PPT1 | Palmitoyl-protein thioesterase 1 | Fatty acid elongation | −7.51 | −2.61 |
| Gh_A01G0045 | KCS9 | 3-ketoacyl-CoA synthase 9 | Fatty acid elongation | 4.63 | 4.02 |
| Gh_A03G0701 | KCS4 | 3-ketoacyl-CoA synthase 4 | Fatty acid elongation | −1.94 | −2.45 |
| Gh_A03G1143 | KCR1 | Very-long-chain 3-oxoacyl-CoA reductase 1 | Fatty acid elongation | 5.43 | 4.23 |
| Gh_A11G3137 | PAS2 | Very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase 2 | Fatty acid elongation | 3.45 | 2.92 |
| Gh_A05G3048 | ECR | Very-long-chain enoyl-CoA reductase | Fatty acid elongation | - | 1.16 |
| Gh_A10G1742 | GPSN2 | Very-long-chain enoyl-CoA reductase | Fatty acid elongation | −4.89 | −2.61 |
| Gh_A01G1527 | LACS2 | Long chain acyl-CoA synthetase 2 | Fatty acid metabolism | 8.41 | 6.67 |
| Gh_A01G0167 | LACS7 | Long chain acyl-CoA synthetase 7, peroxisomal | Fatty acid metabolism | −1.90 | −1.50 |
| Gh_A01G0141 | ACX3 | Acyl-coenzyme A oxidase 3, peroxisomal | Fatty acid metabolism | −2.01 | −1.50 |
| Gh_A02G1616 | ALDH3 | Aldehyde dehydrogenase family 3 | Fatty acid metabolism | 3.67 | 6.33 |
| Gh_D08G2387 | ADH6 | Alcohol dehydrogenase 6 | Fatty acid metabolism | - | −1.37 |
| Gh_A03G1194 | TAA1 | L-tryptophan—pyruvate aminotransferase | Tryptophan metabolism | 5.76 | 3.69 |
| Gh_A05G0035 | YUC4 | Probable indole-3-pyruvate monooxygenase YUCCA4 | Tryptophan metabolism | 4.85 | 7.68 |
| Gh_A08G0657 | YUC10 | Probable indole-3-pyruvate monooxygenase YUCCA10 | Tryptophan metabolism | −4.92 | −2.40 |
| Gh_D09G0992 | AAO2 | Indole-3-acetaldehyde oxidase 2 | Tryptophan metabolism | −2.03 | −2.98 |
| Gh_A02G1509 | CYP | Cytochrome P450 | Tryptophan metabolism | - | −4.14 |
| Gh_A05G2791 | UGT | UDP-glycosyltransferase | Tryptophan metabolism | - | 4.15 |
| Gh_D04G0292 | ALDH3I1 | Aldehyde dehydrogenase family 3 member I1 | Tryptophan metabolism | 1.25 | 2.10 |
| Gh_A01G0390 | PPC | Phosphoenolpyruvate carboxylase | Pyruvate metabolism | 3.86 | 2.07 |
| Gh_D08G2350 | PCKA | Phosphoenolpyruvate carboxykinase (ATP) | Pyruvate metabolism | 1.64 | 1.20 |
| Gh_A10G1783 | PYK | Pyruvate kinase | Pyruvate metabolism | −1.91 | −1.40 |
| Gh_A09G2065 | DLD | Dihydrolipoamide dehydrogenase | Pyruvate metabolism | 2.42 | 2.18 |
| Gh_A05G2129 | DLAT | Dihydrolipoamide acetyltransferase | Pyruvate metabolism | - | 1.26 |
| Gh_A05G3511 | ACSS | Acetyl-CoA synthetase | Pyruvate metabolism | −2.07 | −1.82 |
| Gh_D11G3501 | AAT1 | Acetyl-CoA acetyltransferase, cytosolic 1 | Pyruvate metabolism | - | −3.92 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Guo, H.; Zhang, L.; Fan, Y.; Wu, J.; Tang, Z.; Zhang, Y.; Fan, Y.; Zeng, F. Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Genotype-Recalcitrant Somatic Embryogenesis Transdifferentiation in Cotton. Genes 2020, 11, 519. https://doi.org/10.3390/genes11050519
Guo H, Guo H, Zhang L, Fan Y, Wu J, Tang Z, Zhang Y, Fan Y, Zeng F. Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Genotype-Recalcitrant Somatic Embryogenesis Transdifferentiation in Cotton. Genes. 2020; 11(5):519. https://doi.org/10.3390/genes11050519
Chicago/Turabian StyleGuo, Huihui, Haixia Guo, Li Zhang, Yijie Fan, Jianfei Wu, Zhengmin Tang, Yao Zhang, Yupeng Fan, and Fanchang Zeng. 2020. "Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Genotype-Recalcitrant Somatic Embryogenesis Transdifferentiation in Cotton" Genes 11, no. 5: 519. https://doi.org/10.3390/genes11050519
APA StyleGuo, H., Guo, H., Zhang, L., Fan, Y., Wu, J., Tang, Z., Zhang, Y., Fan, Y., & Zeng, F. (2020). Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Genotype-Recalcitrant Somatic Embryogenesis Transdifferentiation in Cotton. Genes, 11(5), 519. https://doi.org/10.3390/genes11050519