Genome Maintenance by DNA Helicase B


Abstract
:1. Introduction
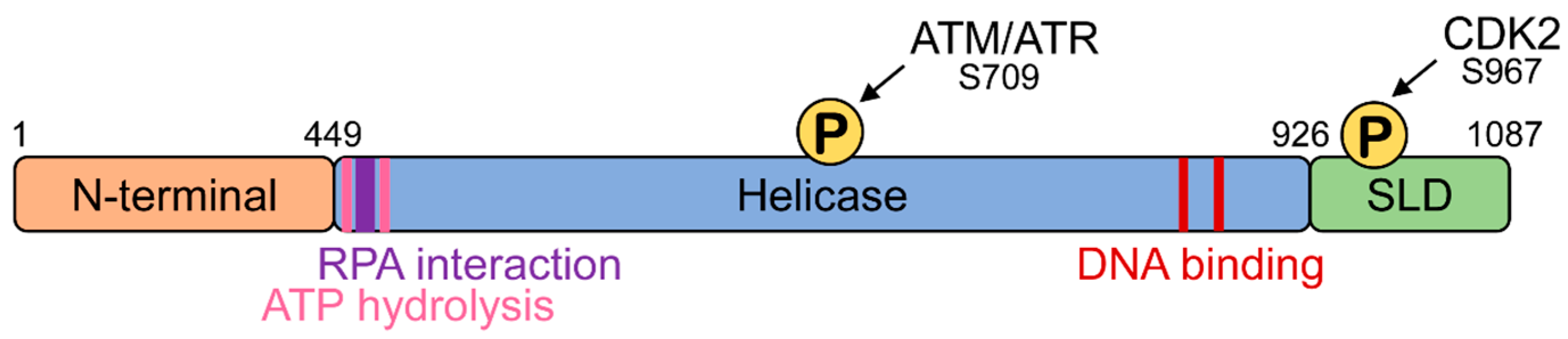
2. Domain Structure
3. Subcellular Localization
4. Functions of Human HELB
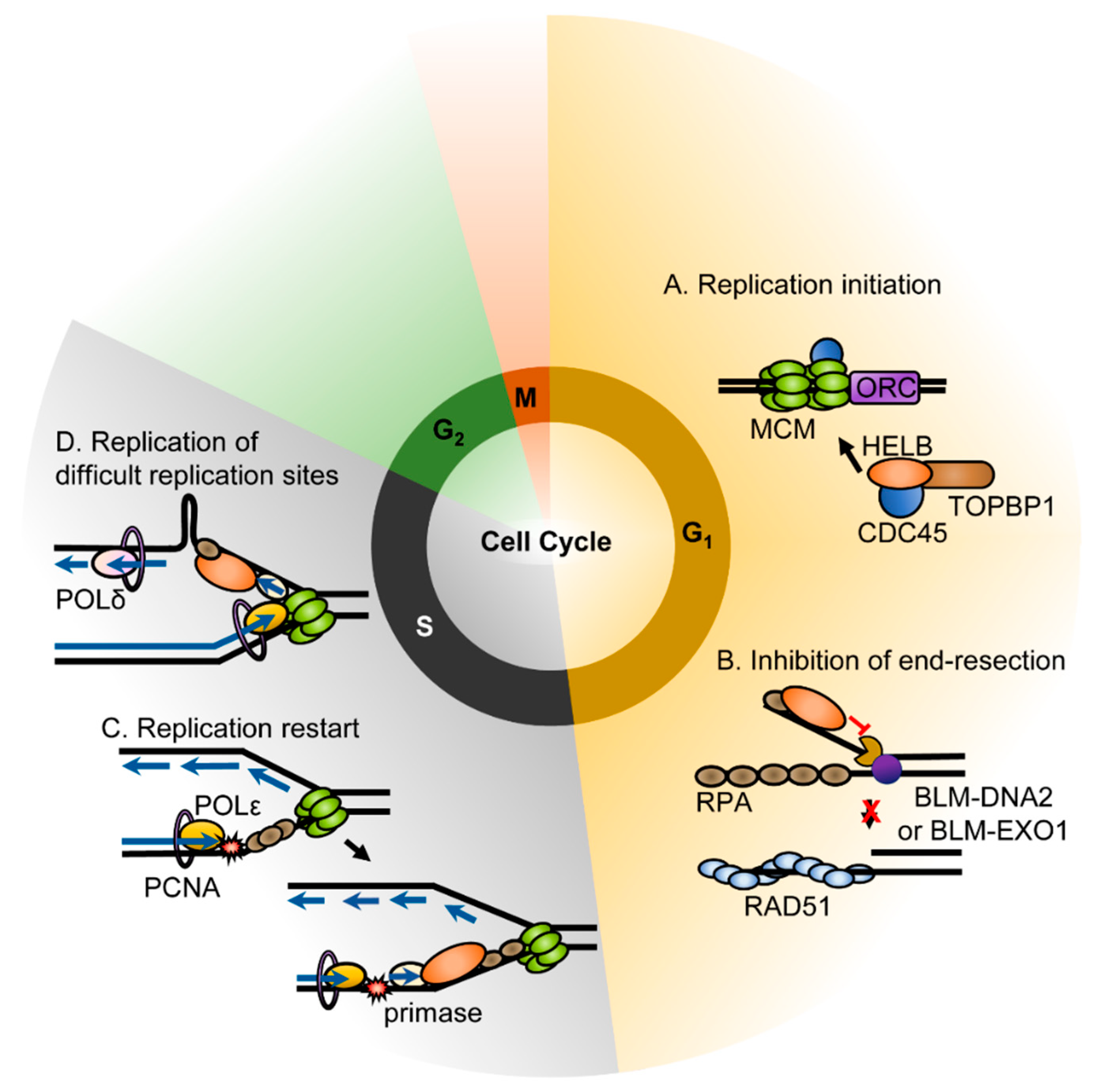
4.1. Role in DNA Replication
4.2. Response to Replication Stress
4.3. Response to DNA Damage
5. Regulation
5.1. Transcriptional Regulation
5.2. Post-Translational Regulation
6. Effects of Variants
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Estep, K.N.; Brosh, R.M. RecQ and Fe–S helicases have unique roles in DNA metabolism dictated by their unwinding directionality, substrate specificity, and protein interactions. Biochem. Soc. Trans. 2018, 46, 77–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneja, P.; Gu, J.; Peng, R.; Carrick, R.; Uchiumi, F.; Ott, R.D.; Gustafson, E.; Podust, V.N.; Fanning, E. A dominant-negative mutant of human DNA helicase B blocks the onset of chromosomal DNA replication. J. Biol. Chem. 2002, 277, 40853–40861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, S.; Kobayashi, T.; Omori, A.; Kusa, Y.; Okumura, N.; Kodaira, H.; Ishimi, Y.; Seki, M.; Enomoto, T. Molecular cloning of a cDNA encoding mouse DNA helicase B, which has homology to Escherichia coli RecD protein, and identification of a mutation in the DNA helicase B from tsFT848 temperature-sensitive DNA replication mutant cells. Nucleic Acids Res. 2001, 29, 3835–3840. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Kohda, T.; Yano, T.; Tada, S.; Yanagisawa, J.; Eki, T.; Ui, M.; Enomoto, T. Characterization of DNA synthesis and DNA-dependent ATPase activity at a restrictive temperature in temperature-sensitive tsFT848 cells with thermolabile DNA helicase B. Mol. Cell. Biol. 1995, 15, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, A.; Tada, S.; Katada, T.; Enomoto, T. Stimulation of mouse DNA primase-catalyzed oligoribonucleotide synthesis by mouse DNA helicase B. Nucleic Acids Res. 1995, 23, 2014–2018. [Google Scholar] [CrossRef]
- Tkac, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Diaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xia, X.; Yan, P.; Liu, H.; Podust, V.N.; Reynolds, A.B.; Fanning, E. Cell cycle-dependent regulation of a human DNA helicase that localizes in DNA damage foci. Mol. Biol. Cell 2004, 15, 3320–3332. [Google Scholar] [CrossRef]
- Gerhardt, J.; Guler, G.D.; Fanning, E. Human DNA helicase B interacts with the replication initiation protein Cdc45 and facilitates Cdc45 binding onto chromatin. Exp. Cell Res. 2015, 334, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Fairman-Williams, M.E.; Guenther, U.P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Guler, G.D.; Liu, H.; Vaithiyalingam, S.; Arnett, D.R.; Kremmer, E.; Chazin, W.J.; Fanning, E. Human DNA helicase B (HDHB) binds to replication protein A and facilitates cellular recovery from replication stress. J. Biol. Chem. 2012, 287, 6469–6481. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.T.; Jones, J.T.; Meyer, T. Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle 2009, 8, 1044–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, N.K.; Cordin, O.; Banroques, J.; Doere, M.; Linder, P. The Q Motif: A Newly Identified Motif in DEAD Box Helicases May Regulate ATP Binding and Hydrolysis. Mol. Cell 2003, 11, 127–138. [Google Scholar] [CrossRef]
- Shen, Z.; Prasanth, S.G. Emerging players in the initiation of eukaryotic DNA replication. Cell Div. 2012, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Guler, G.D.; Rosenwaks, Z.; Gerhardt, J. Human DNA Helicase B as a Candidate for Unwinding Secondary CGG Repeat Structures at the Fragile X Mental Retardation Gene. Front. Mol. Neurosci. 2018, 11, 138. [Google Scholar] [CrossRef]
- Fry, M.; Loeb, L.A. Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem. 1999, 274, 12797–12802. [Google Scholar] [CrossRef] [Green Version]
- Marquis Gacy, A.; Goellner, G.; Juranić, N.; Macura, S.; McMurray, C.T. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 1995, 81, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef] [Green Version]
- Samadashwily, G.M.; Raca, G.; Mirkin, S.M. Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 1997, 17, 298–304. [Google Scholar] [CrossRef]
- Burrow, A.A.; Marullo, A.; Holder, L.R.; Wang, Y.-H. Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res. 2010, 38, 2865–2877. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Freudenreich, C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosom. Cancer 2018, 58, gcc.22721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Brison, O.; El-Hilali, S.; Azar, D.; Koundrioukoff, S.; Schmidt, M.; Nähse, V.; Jaszczyszyn, Y.; Lachages, A.M.; Dutrillaux, B.; Thermes, C.; et al. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun. 2019, 10, 5693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliam, T.A.; Doherty, A.J. Primpol—Prime time to reprime. Genes 2017, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, L. The Pol α -Primase Complex. Subcell. Biochem. 2012, 62, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Chaudhuri, A.R.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Sung, P. To Cut or Not to Cut: Discovery of a Novel Regulator of DNA Break Resection. Mol. Cell 2016, 61, 325–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Tutt, A.N.J.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; Van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Ross Chapman, J.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Uchiumi, F.; Arakawa, J.; Iwakoshi, K.; Ishibashi, S.; Tanuma, S. Characterization of the 5’-flanking region of the human DNA helicase B (HELB) gene and its response to trans-Resveratrol. Sci. Rep. 2016, 6, 24510. [Google Scholar] [CrossRef]
- Gehm, B.D.; McAndrews, J.M.; Chien, P.Y.; Jameson, J.L. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 14138–14143. [Google Scholar] [CrossRef] [Green Version]
- Petz, L.N.; Nardulli, A.M. Sp1 Binding Sites and An Estrogen Response Element Half-Site Are Involved in Regulation of the Human Progesterone Receptor A Promoter. Mol. Endocrinol. 2000, 14, 972–985. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Visser, J.A.; Zhu, J.; Burrows, C.J. Human DNA Repair Genes Possess Potential G-Quadruplex Sequences in Their Promoters and 5’-Untranslated Regions. Biochemistry 2018, 57, 991–1002. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Kendrick, S.; Hecht, S.M.; Hurley, L.H. The transcriptional complex between the BCL2 i-motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 2014, 136, 4172–4185. [Google Scholar] [CrossRef] [PubMed]
- Assi, H.A.; Garavís, M.; González, C.; Damha, M.J. I-motif DNA: Structural features and significance to cell biology. Nucleic Acids Res. 2018, 46, 8038–8056. [Google Scholar] [PubMed] [Green Version]
- Kim, N. The Interplay between G-quadruplex and Transcription. Curr. Med. Chem. 2017, 26, 2898–2917. [Google Scholar] [CrossRef]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [Green Version]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (k-ε-gg) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. Ubisite approach for comprehensive mapping of lysine and n-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [Green Version]
- García-Rodríguez, N.; Wong, R.P.; Ulrich, H.D. Functions of ubiquitin and SUMO in DNA replication and replication stress. Front. Genet. 2016, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, F.R.; Ruth, K.S.; Thompson, D.J.; Lunetta, K.L.; Pervjakova, N.; Chasman, D.I.; Stolk, L.; Finucane, H.K.; Sulem, P.; Bulik-Sullivan, B.; et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 2015, 47, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, D.A.; Kane, R.L.; Beeson, W.L.; Burke, G.L.; Sprafka, J.M.; Potter, J.; Iso, H.; Jacobs, D.R.; Phillips, R.L. Is early natural menopause a biologic marker of health and aging? Am. J. Public Health 1989, 79, 709–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richelson, L.S.; Wahner, H.W.; Melton, L.J.; Riggs, B.L. Relative Contributions of Aging and Estrogen Deficiency to Postmenopausal Bone Loss. N. Engl. J. Med. 1984, 311, 1273–1275. [Google Scholar] [CrossRef]
- Kelsey, J.L.; Gammon, M.D.; John, E.M. Reproductive Factors and Breast Cancer. Epidemiol. Rev. 1993, 15, 36–47. [Google Scholar] [CrossRef]
- Lambalk, C.B.; van Disseldorp, J.; de Koning, C.H.; Broekmans, F.J. Testing ovarian reserve to predict age at menopause. Maturitas 2009, 63, 280–291. [Google Scholar] [CrossRef]
- Hunter, N. Meiotic recombination: The essence of heredity. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| SNP | Alleles | Variant | MAF | Position (Chr:12) | P-Value |
|---|---|---|---|---|---|
| rs12301608 | C/T | Intronic | 0.0125 | 66,707,644 | 2.24 × 10−5 |
| rs12228262 | G/C | Intronic | 0.0125 | 66,708,507 | 2.28 × 10−5 |
| rs10878404 | C/T | Intronic | 0.0122 | 66,709,488 | 1.74 × 10−5 |
| rs76187362 | A/G | Intronic | 0.0125 | 66,711,895 | 2.47 × 10−5 |
| rs79976130 | C/T | Intronic | 0.0125 | 66,712,652 | 2.52 × 10−5 |
| rs10878406 | T/C | Intronic | 0.0122 | 66,713,978 | 3.63 × 10−5 |
| rs10878407 | C/T | Intronic | 0.0124 | 66,717,202 | 5.38 × 10−5 |
| rs35536133 | T/A | Exonic/synonymous | 0.0124 | 66,717,784 | 6.06 × 10−5 |
| rs28551050 | G/T | Intronic | 0.0124 | 66,718,207 | 6.62 × 10−5 |
| rs10878408 | C/G | Intronic | 0.0122 | 66,718,973 | 6.31 × 10−5 |
| rs139815108 | C/T | Intronic | 0.0113 | 66,718,957 | 3.44 × 10−5 |
| rs34109029 | G/T | Intronic | 0.0124 | 66,717,910 | 6.29 × 10−5 |
| rs60549090 | G/T | Intronic | 0.0122 | 66,705,808 | 1.63 × 10−5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hazeslip, L.; Zafar, M.K.; Chauhan, M.Z.; Byrd, A.K. Genome Maintenance by DNA Helicase B. Genes 2020, 11, 578. https://doi.org/10.3390/genes11050578
Hazeslip L, Zafar MK, Chauhan MZ, Byrd AK. Genome Maintenance by DNA Helicase B. Genes. 2020; 11(5):578. https://doi.org/10.3390/genes11050578
Chicago/Turabian StyleHazeslip, Lindsey, Maroof Khan Zafar, Muhammad Zain Chauhan, and Alicia K. Byrd. 2020. "Genome Maintenance by DNA Helicase B" Genes 11, no. 5: 578. https://doi.org/10.3390/genes11050578
APA StyleHazeslip, L., Zafar, M. K., Chauhan, M. Z., & Byrd, A. K. (2020). Genome Maintenance by DNA Helicase B. Genes, 11(5), 578. https://doi.org/10.3390/genes11050578